
Every quality team has a deviation process on paper. Most of them look reasonable in an SOP. And yet, deviation backlogs keep forming. Extensions keep getting granted, investigations sit open for weeks past their deadlines, and the queue grows faster than the team can close it.
Sound familiar?
The gap between what's documented and what actually happens on the floor is where deviation programs break down. And when they break down, the consequences compound quickly: regulatory exposure, delayed CAPAs, repeat deviations that never get their root cause addressed, and a quality team that spends all its time firefighting instead of preventing fires.
We've embedded consultants into manufacturing operations where the deviation log had swollen to 80 or more open events at a time, where extensions were being handed out routinely, and where regulatory authorities had taken notice.
What follows is a practical framework for deviation management drawn from those engagements — not theory, but a model that was built, tested, refined through some false starts, and ultimately adopted by senior leadership as the facility's standard operating approach.
The specifics will vary from site to site, but the structural elements are transferable to any GMP manufacturing environment. Use it as a playbook to adapt into a system of your own.
Start by Diagnosing Why the Backlog Exists
Before you redesign anything, you need to understand where time is actually being lost.
The instinct when staring down a large backlog is to start processing — just get people writing and closing. That instinct is wrong, or at least incomplete, because if you don't address the systemic reasons the backlog formed in the first place, you will rebuild it.

In the engagement from which this framework originated, the root causes were not mysterious or exotic. They were mundane and structural:
If any of these sound familiar, they should! They're the same five or six problems that drive deviation backlogs at most manufacturing sites. The specifics differ of course, but the pattern is remarkably consistent.
The Framework: Stage Gates, Daily Accountability, and Information Flow
The deviation management model that emerged from this engagement was not designed in a conference room and rolled out fully formed.
It evolved naturally over months, went through false starts, and was refined as the team learned what worked and what didn't. It was eventually formalized by senior leadership and adopted as the site's standard approach.
Here are its core elements, visualized first and then explained.
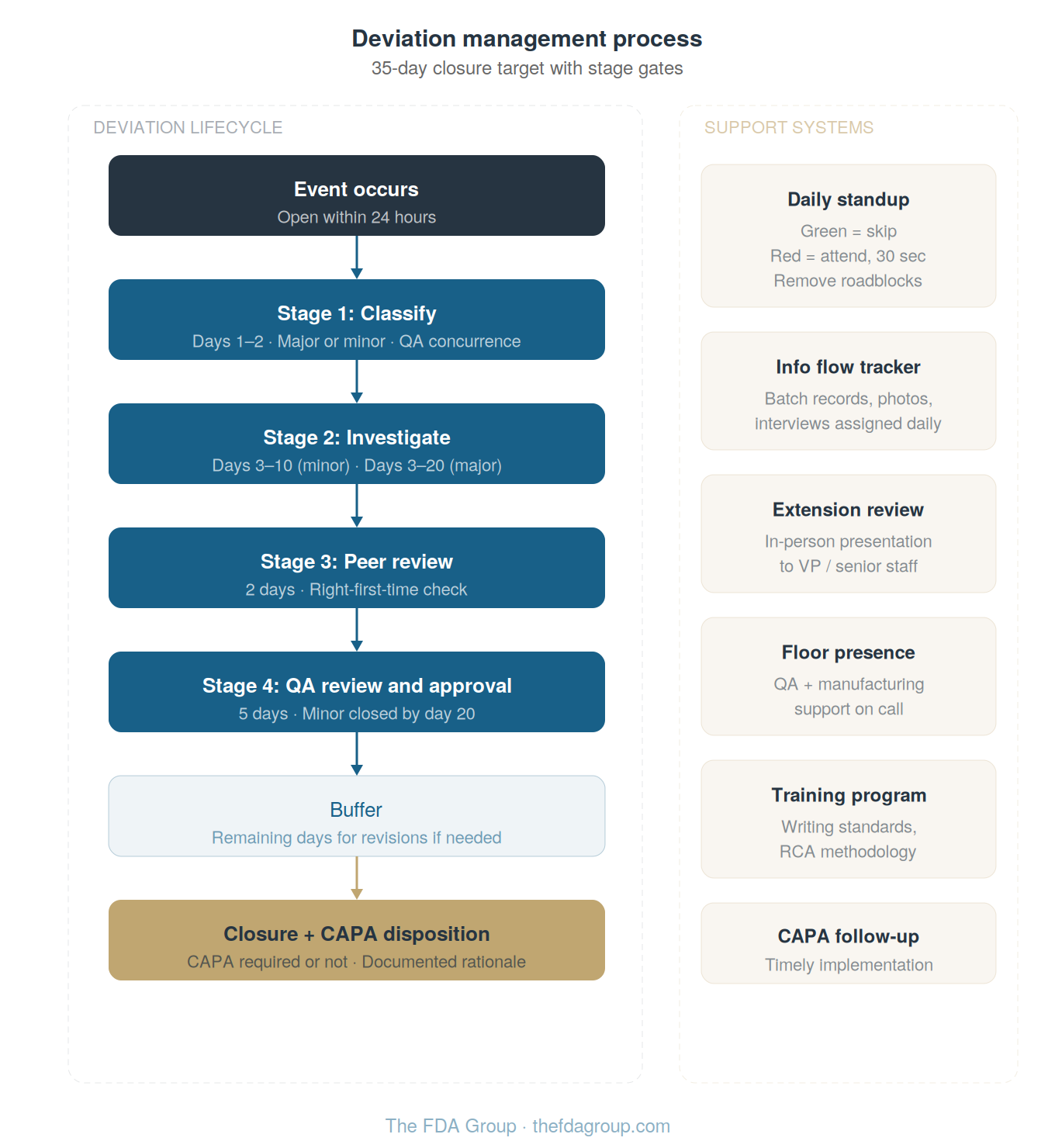
Define your timeline and break it into "stage gates"
The facility settled on a 35-day closure target for all deviations. This is generally applicable to just about any site and situation. Within that window, the process was broken into defined stages, each with a specific deadline:
Keep in mind that these specific numbers are not sacred. Your site may need a 30-day window or a 45-day window. The stage gates may fall on different days.
What matters is that you define them, publish them, and hold people to them. The absence of defined timelines is what allows deviations to sit indefinitely. And once one sits, they all start sitting.
Establish a daily standup meeting
This is maybe the single most important operational element in the framework, and the one most likely to face resistance.
Here is how it works:
A few practical notes from our experience running this process:
- The meeting must be short. If it runs longer than 15 to 20 minutes, something is wrong with how it's being run. Red items only. Thirty seconds each. Assign actions and move on.
-
Management must attend and must follow through. In the early iterations of this meeting, actions were assigned, but nobody tracked follow-up. The spreadsheet said someone was supposed to get a batch record to the writer. Three days later, the writer still did not have it. The meeting only works if the actions it generates actually happen. Daily accountability means daily, not "I'll get to it."
- Senior leadership buy-in is non-negotiable. The meeting didn't gain real traction until the site director mandated it and began personally reviewing the metrics. If it is treated as a mid-level quality exercise, it will be treated as optional by the support functions whose cooperation is essential.
Fix information flow: it's probably your biggest time sink
If you take one thing from this guide, let it be this:
The single biggest reason deviation timelines slip is that writers cannot get the information they need to start investigating.
In a 35-day timeline, burning five to seven days just assembling batch records, photos, operator statements, and equipment logs means you have lost 15 to 20 percent of your window before any substantive work begins. That time loss is invisible in most deviation-tracking systems because it falls under the generic umbrella of "investigation," but it's not investigation—it's logistics!
The fix is structural, not motivational. You can't tell writers to "be more proactive" about getting information. You need a system that actually surfaces information gaps daily and assigns responsibility for filling them.
The daily meeting does this if it is run correctly. A shared spreadsheet or tracker where writers log what they need (specific batch records, specific interview subjects, specific equipment logs) makes those needs visible to management. Assigning a named person to each request, with a due date, creates accountability. Reviewing those assignments the following day closes the loop.
This also means the people who open deviations need to provide adequate initial documentation. If the initial deviation report is a one-line description with no supporting detail, the writer is starting from zero. Training the people who initiate deviations — often manufacturing operators or supervisors — to include basic information (what happened, when, what equipment or batch was involved, whether product is potentially affected, photos if relevant) can save days per deviation downstream.
Make extensions painful
Extensions are the pressure relief valve that allows deviation programs to fail quietly. If extensions are easy to get, deadlines are suggestions. If deadlines are suggestions, timelines drift. If timelines drift, the backlog grows.
The most effective mechanism we have seen: require anyone requesting an extension to present their case to the VP or site director in person. Not an email. Not a form. A brief, formal presentation explaining why the deviation cannot be closed within the allotted time and what specific steps remain.
Waiting for external lab results or stability data from an outside facility? That is a legitimate reason. "I didn't have time" or "I was busy with other things" is not.
The goal is not to be punitive. The goal is to create enough friction around extensions that people find ways to avoid needing them. When extensions were handed out freely at this facility, they were everywhere. Once the in-person review was implemented, the number dropped dramatically.
Put QA and manufacturing support on the floor
Most deviation programs we see are inherently reactive. Something goes wrong, a deviation gets opened, and an investigation happens after the fact. The most impactful preventive measure in this framework was putting QA and manufacturing support personnel on call on the production floor during operations.
When an operator had a question, someone was there to answer it, before the question became a deviation. When something went wrong, manufacturing support was there immediately, preventing a small issue from escalating. The consultant who helped build this program described it as having smoke detectors going off early, before a small problem becomes a fire.
Not everyone liked this at first! Floor presence felt like oversight to some operators and supervisors. But the reduction in avoidable deviations (missed signatures, documentation errors, process missteps that could have been caught in real time) quickly justified it.

Writing Deviations that Actually Close
A deviation program can have perfect stage gates, perfect daily meetings, and perfect information flow, and still bog down if the investigations themselves are poorly written. QA sends them back. Inspectors start pulling on loose threads to find more problems. The same deviation gets reworked two or three times.
Here are the writing principles that made the biggest difference in the engagement this framework comes from:
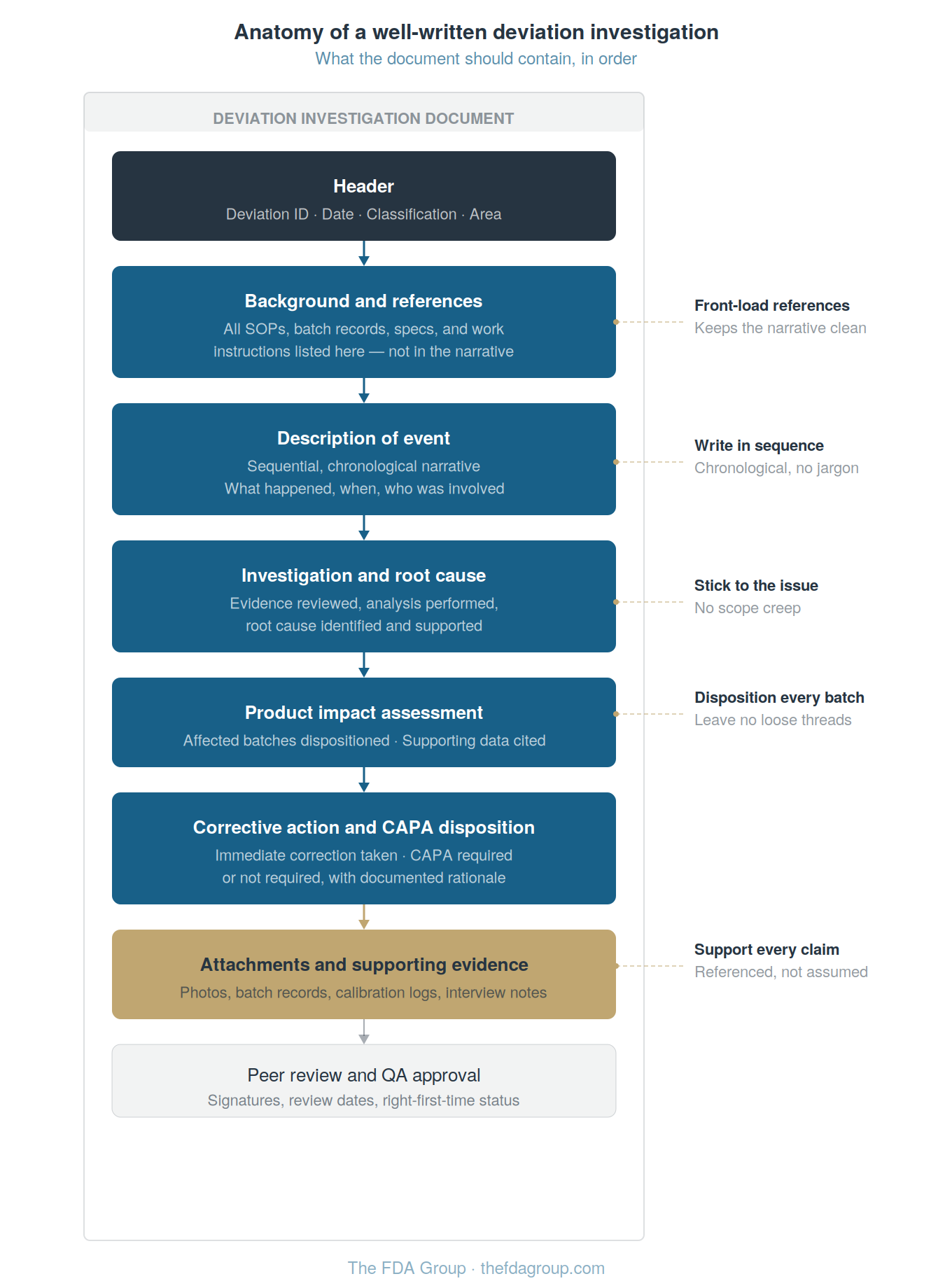
1. Write for the reader who knows nothing about the process
The person reviewing your investigation, whether it's a QA reviewer or an FDA inspector, may have no context for what happened. Your narrative needs to educate them. If someone can pick up the investigation cold and understand what occurred, what was affected, and what was done about it, you have written it correctly.
2. Tell the story in sequence
At 10:45, this happened. The operator contacted the QA supervisor. The supervisor directed that valves two and three be shut off. The spill was approximately two liters. See attachment one for photos. The batch record was annotated.
A linear, chronological narrative is the clearest way to communicate what happened and is the easiest for a reviewer to follow.
3. Front-load your references
Many deviation writers bury SOP citations and document references throughout the narrative, which makes the investigation painful to read.
Instead, list all relevant documents (SOPs, batch records, specifications, work instructions) in a background section at the top. Then reference them by number in the narrative. The reader can look up any reference if they need context, but the narrative itself stays clean and readable.
4. Stick to the issue
Scope creep is the enemy of timely closure. Address the specific deviation event: what happened, why, whether product was affected, and what corrective action is needed. If the investigation reveals a broader systemic issue — say, inconsistent visual inspection practices across the facility — flag it and recommend addressing it through a separate improvement project.
Do not try to solve every upstream problem within a single deviation investigation. That is how investigations become impossible to close.
5. Leave no loose threads
Every claim should be supported. Every affected batch should be dispositioned. Every question a reviewer might ask should be answered in the document.
One consultant described the ideal deviation investigation as a sphere: completely smooth, nothing sticking out for someone to grab and pull on. An inspector who picks up a well-written investigation and finds no loose ends moves on. An inspector who finds a thread pulls it, and that pull can unravel far more than the single deviation.
Assign Deviations by Expertise, not Availability
Not all deviations require the same technical knowledge.
Equipment failures are different from chemistry-related process deviations, which are different from documentation errors. Assigning deviations based on who has capacity (rather than who has relevant expertise) slows investigations, increases rework, and frustrates everyone involved.
Here's a better approach: let writers volunteer for deviations they are qualified to handle. People with equipment and engineering backgrounds take the equipment deviations. People with process chemistry expertise take the chemistry deviations. People who are fast, thorough writers take the high-volume minor deviations that require less technical depth but still consume significant time.
This sounds obvious, but many deviation programs assign work by rotation or by whoever has the fewest open items, regardless of fit. Matching expertise to event type reduces investigation time, improves quality, and reduces the rate at which QA sends investigations back for revision.
Don't Forget the CAPA Downstream
Clearing a deviation backlog generates CAPA work. Every deviation that identifies a root cause requiring corrective action creates a CAPA obligation. If you resource for deviation closure but not for the CAPA work that follows, you've not solved the problem; you've moved the bottleneck downstream.
Plan for it!
-
Track CAPA initiation and follow-up alongside your deviation metrics.
-
Hold CAPA implementation to the same stage gate discipline and daily accountability you apply to deviations.
The organizations that break the backlog cycle are the ones that close the loop all the way through CAPA verification of effectiveness, not just through deviation closure.

The Elements That Matter Most
If you're looking at your own deviation program and wondering where to start, here is the short list in priority order:
None of this is revolutionary, and that's the point! Deviation management breaks down not because the concepts are complicated but because the execution requires discipline, accountability, and sustained management attention. The framework above provides the structure. The discipline has to come from leadership.
This framework is drawn from a staff augmentation engagement in which The FDA Group embedded a senior quality consultant into a biologics manufacturing operation for nearly two years. For more on the engagement itself, see our companion case study. If your deviation program needs structural improvement or your team needs experienced resources to help implement a framework like this, reach out to us.