
Updated: February 2023
This guide serves as an ongoing report of the most recent FDA inspection and enforcement trends, specifically in the area of good manufacturing practice (GMP), based on publicly available data. We've included a mix of our firsthand research along with others' analyses and links to the appropriate sources.
Note that the data presented here conform to the fiscal year accounting period for the federal government, which begins on October 1 and ends on September 30. The fiscal year is designated by the calendar year in which it ends.
We've kept trending reports from previous years, as changes from year to year tend to be subtle. Past figures still provide extremely valuable insight teams can use to assess their own state of compliance and take action if improvements are necessary.
Need expert help planning and executing compliance projects or connecting with the industry's top quality and compliance professionals? Get in touch with us and learn more about how we help 17 of the top 25 life science companies execute their projects on time and on budget.
Use the links below to jump to a specific section.
- 2023 Trend Highlights
- Top 5-Year Cumulative Trending FDA GMP Inspection Citations (FY2018-FY2022)
- Major Takeaways from The State of Pharmaceutical Quality: Fiscal Year 2021
- Major Takeaways from The State of Pharmaceutical Quality: Fiscal Year 2020
- Pre-Pandemic FDA Inspection and Enforcement Trends
- Major Takeaways from The State of Pharmaceutical Quality: Fiscal Year 2019
- Takeaways, Resources, and Next Steps
- Get Expert Quality & Compliance Assistance Now
FDA Warning Letter & Inspection Trend Highlights: 2023
|
Top 5-Year (FY2018-FY2022) Cumulative Trending FDA Inspection Citations (GMP)
The following trend data has been analyzed from data gathered from FDA's data dashboard Inspection Citation datasets.
- Citations data include Form FDA 483 citations and may not necessarily represent citations on final classification letters.
- Also, we've filtered this data to only report on the primary GMP regulations:
- Drugs — 21 CFR Part 210 and 21 CFR Part 211
- Medical Devices — 21 CFR Part 820 and 21 CFR 803.17
Please note that there are important caveats FDA makes clear on its dashboard page regarding the completeness of the data it includes.
- For example, not all inspections are included in the database. Inspections conducted by states, pre-approval inspections, mammography facility inspections, inspections waiting for a final enforcement action, and inspections of nonclinical labs are not included.
- Inspections of nonclinical labs are available at Nonclinical Laboratories Inspected under Good Laboratory Practices.
- Read all of FDA's data dashboard caveats here.
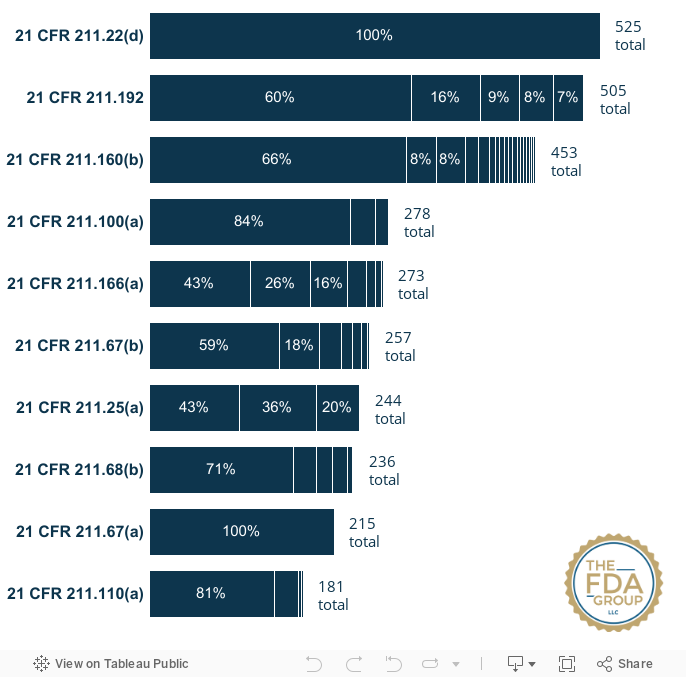
Top 10 GMP Drug Inspections Citations: FY2018 - FY2022
Citations data include Form FDA 483 citations and may not necessarily represent citations on final classification letters.
| Act/CFR Number | Number of Citations FY2018-FY2022 |
|
#1 — 21 CFR 211.22(d) — The responsibilities and procedures applicable to the quality control unit shall be in writing; such written procedures shall be followed |
525 |
|
#2 — 21 CFR 211.192 — Production record review |
505 |
|
#3 — 21 CFR 211.160(b) — Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedures... |
453 |
| #4 — 21 CFR 211.100(a) — There shall be written procedures for production and process control designed to assure that the drug products have the identity, strength, quality, and purity they purport or are represented to possess... | 278 |
|
#5 —21 CFR 211.166(a) — There shall be a written testing program designed to assess the stability characteristics of drug products. The results of such stability testing shall be used in determining appropriate storage conditions and expiration dates... |
273 |
|
#6 — 21 CFR 211.67(b)— Written procedures shall be established and followed for cleaning and maintenance of equipment, including utensils, used in the manufacture, processing, packing, or holding of a drug product... |
257 |
| #7 — 21 CFR 211.25(a) — Each person engaged in the manufacture, processing, packing, or holding of a drug product shall have education, training, and experience, or any combination thereof, to enable that person to perform the assigned functions... | 244 |
| #8 — 21 CFR 211.68(b) — Appropriate controls shall be exercised over computer or related systems to assure that changes in master production and control records or other records are instituted only by authorized personnel... | 236 |
| #9 — 21 CFR 211.67(a) — Equipment and utensils shall be cleaned, maintained, and, as appropriate for the nature of the drug, sanitized and/or sterilized at appropriate intervals to prevent malfunctions or contamination that would alter the safety, identity, strength, quality, or purity of the drug product beyond the official or other established requirements. | 215 |
| #10 — 21 CFR 211.110(a) — To assure batch uniformity and integrity of drug products, written procedures shall be established and followed that describe the in-process controls, and tests, or examinations to be conducted on appropriate samples of in-process materials of each batch. Such control procedures shall be established to monitor the output and to validate the performance of those manufacturing processes that may be responsible for causing variability in the characteristics of in-process material and the drug product... | 181 |
Given that FDA's inspectional citations specify different descriptions and particular subparts, the interactive chart below breaks down these details to show the relative prevalence of certain observed issues over others.
Hover over each section to see the specific short description given under each CFR reference number/subpart.
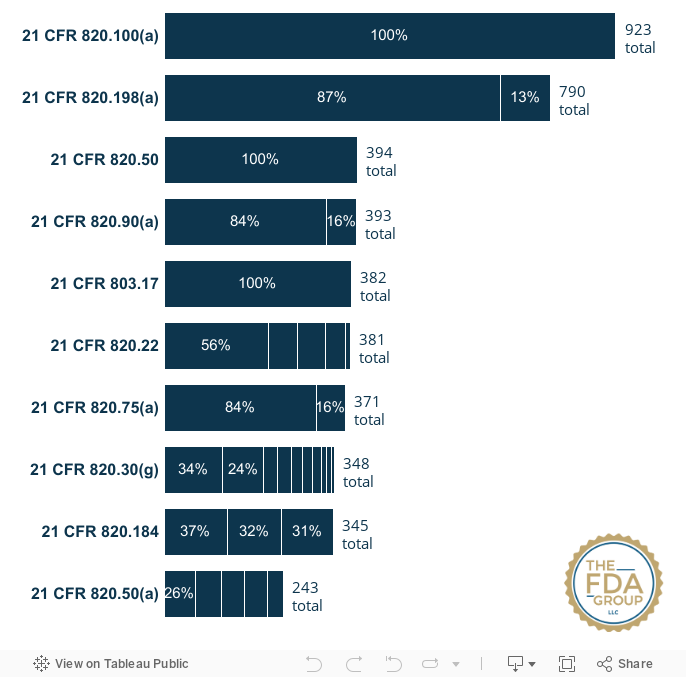
Top 10 GMP Medical Device Inspections Citations: FY2018 - FY2022
Citations data include Form FDA 483 citations and may not necessarily represent citations on final classification letters.
| Act/CFR Number | Number of Citations FY2018-FY2022 |
|
#1 — 21 CFR 820.100(a) — Each manufacturer shall establish and maintain procedures for implementing corrective and preventive action. |
923 |
|
#2 — 21 CFR 820.198(a) — Each manufacturer shall maintain complaint files. Each manufacturer shall establish and maintain procedures for receiving, reviewing, and evaluating complaints by a formally designated unit... |
790 |
|
#3 — 21 CFR 820.50 — Each manufacturer shall establish and maintain procedures to ensure that all purchased or otherwise received product and services conform to specified requirements. |
394 |
| #4 — 21 CFR 820.90(a) — Each manufacturer shall establish and maintain procedures to control product that does not conform to specified requirements... | 393 |
| #5 — 21 CFR 803.17 — If you are a user facility, importer, or manufacturer, you must develop, maintain, and implement written MDR procedures... | 382 |
|
#6 — 21 CFR 820.22 — Each manufacturer shall establish procedures for quality audits and conduct such audits to assure that the quality system is in compliance with the established... |
381 |
| #7 — 21 CFR 820.75(a) — Where the results of a process cannot be fully verified by subsequent inspection and test, the process shall be validated with a high degree of assurance and approved according to established procedures... | 371 |
| #8 — 21 CFR 820.30(g) — Design validation. Each manufacturer shall establish and maintain procedures for validating the device design... | 348 |
| #9 — 21 CFR 820.184 — Each manufacturer shall maintain device history records (DHRs)... | 345 |
| #10 — 21 CFR 820.50(a) — Evaluation of suppliers, contractors, and consultants. Each manufacturer shall establish and maintain the requirements, including quality requirements, that must be met by suppliers, contractors, and consultants... | 243 |
Given that FDA's inspectional citations specify different descriptions and particular subparts, the interactive chart below breaks down these details to show the relative prevalence of certain observed issues over others.
Hover over each section to see the specific short description given under each CFR reference number/subpart.
Need regulatory compliance support in 2023?Learn more about our flexible staff augmentation (full-time consultant), FTE recruitment, and consulting services, including GxP auditing and remediation support. We also offer a number of free white papers related to solving quality and compliance problems in some of these trending areas:
|
Free webinar: Identifying and Preventing the Most Common Drug and Device cGMP Issues
In 2021, we hosted a free, two-hour webinar event with accomplished RA/QA consultant Neal Siegel, who addressed many of the top trending drug and device citations year to year and offered firsthand insight into proactive motions teams should consider to ensure compliance. Download the full video and slide deck here.
Webinar Table of Contents
Use the following timecodes to jump to a specific section.
Drug:
- 4:55 —211.192 — Investigations of discrepancies
- 39:41 — 211.22 — Responsibilities of the quality control unit
- 43:16 — 211.160(b) — Lab controls (scientifically sound specifications)
- 54:06 — 211.100(a) — Written procedures for production and process controls
Device:
- 1:00:16 — 820.100(a) — CAPA
- 1:12:21 — 820.198(a) — Complaint files
- 1:27:16 — 820.90(a) — Control of nonconforming product
- 1:36:46 — 820.75 — Process validation
Major Takeaways from the Report on the State of Pharmaceutical Quality: Fiscal Year 2021
In 2022, the Office of Pharmaceutical Quality (OPQ) within FDA’s Center for Drug Evaluation and Research (CDER) published its fiscal year 2021 report on the state of pharmaceutical quality. We distilled some of its main takeaways below.
Read the full report (PDF) on fda.gov here.
- OPQ said its New Inspection Protocol Project (NIPP) program has increased the efficiency of inspections through a more targeted and data-driven approach to identify potential quality problems early on. FDA says its NIPP has “improved how data from pre-approval and surveillance inspections are evaluated and reported.” (FDA has been using these inspection protocols for certain sterile surveillance and pre-approval inspections since 2018.)
- Sites making “essential medicines” that protect the public against outbreaks of emerging diseases such as COVID-19 have high median site inspection scores (SIS), indicating a high rate of compliance with GMP. An analysis of active pharmaceutical ingredient (API) and finished dosage form (FDF) sites found that the median SIS for essential medicine manufacturers was 7.45 out of 10, a score that was “significantly higher” than the 7.0 score for non-essential medicine manufacturers. “This observation indicates that sites manufacturing EM products have a higher level of adherence to manufacturing compliance standards than sites that do not manufacture EM products,” OPQ wrote.
- For the second year, the number of total recalls (particularly Class I recalls) has increased. This follows a three-year period of declining recalls from FY2017 to FY2019. Since FY2016, recalls spiked up dramatically, going from roughly 300 recalls events a year in 2019 to 700 in FY2020 to 800 in FY2021. Hand sanitizers that contained methanol, as well as consumer products and sunscreens with benzene contamination are largely to blame for the increase.
- Roughly half (49.1%) of 1,143 eligible firms did not submit field alert reports (FARs) to the agency over a four-year period from FY2018 to FY2021. FDA’s postmarket reporting requirements specify that sites submit FARs after receiving information on significant quality problems with their distributed drug products.
- A growing number of products are failing sampling and testing requirements; a method of inspection is used when FDA cannot get to sites to conduct inspections. In FY2021, the percentage of non-compliant samples grew to 35%, an increase from 16% in FY2020. The growing rate of non-compliance “is driven by focused sampling assignments with high non-compliant rates for products with nitrosamine contamination, hand sanitizers, and sampling related to COVID-19 mission critical sampling and testing, which became more prominent in FY2021.“
Major Takeaways from the Report on the State of Pharmaceutical Quality: Fiscal Year 2020
Read FDA's full FY2022 report (PDF) on fda.gov here. We distilled some of its main takeaways below. Read our blog post for more depth into some of these takeaways.
- The total number of drug manufacturing sites in CDER’s Site Catalog decreased by 1.2%. Excluding medical gas manufacturers and newly registered hand sanitizer manufacturers, the FY2020 CDER Site Catalog has 4,221 drug manufacturing sites, a small decrease (-1.2%) from 4,273 in FY2019.
- FDA used Mutual Recognition Agreements (MRAs) and its authority to obtain records for sites in advance or in lieu of inspections due to the pandemic. In 2012, the Food and Drug Administration Safety and Innovation Act gave FDA new authorities under the Food, Drug, and Cosmetic Act §704(a)(4) to request records or other information from firms in advance of or in lieu of an inspection.
- “FDA surveillance history, requests for records, and inspection reports obtained through the MRAs were all used to mitigate risk and enable regulatory actions.”
- “As a result of utilizing all of these approaches, CDER completed facility assessments to meet User Fee dates over 90% of the time and reduced the need to conduct PAIs 58% of the time in FY2020 Q3, and 64% of the time in FY2020 Q4."
- “MRA authority was used to assess 183 sites through MRA inspection reports for a total of 745 sites (18% of the FY2020 CDER Site Catalog). For comparison, in FY2019 1,258 drug quality assurance inspections were performed and an additional 109 sites were assessed using MRAs for a total of 1,367 sites (32% of the FY2020 CDER Site Catalog).”
- The number of Warning Letters issued in FY2020 was slightly lower than in FY2018 or FY2019, but still over four times higher than in FY2015.
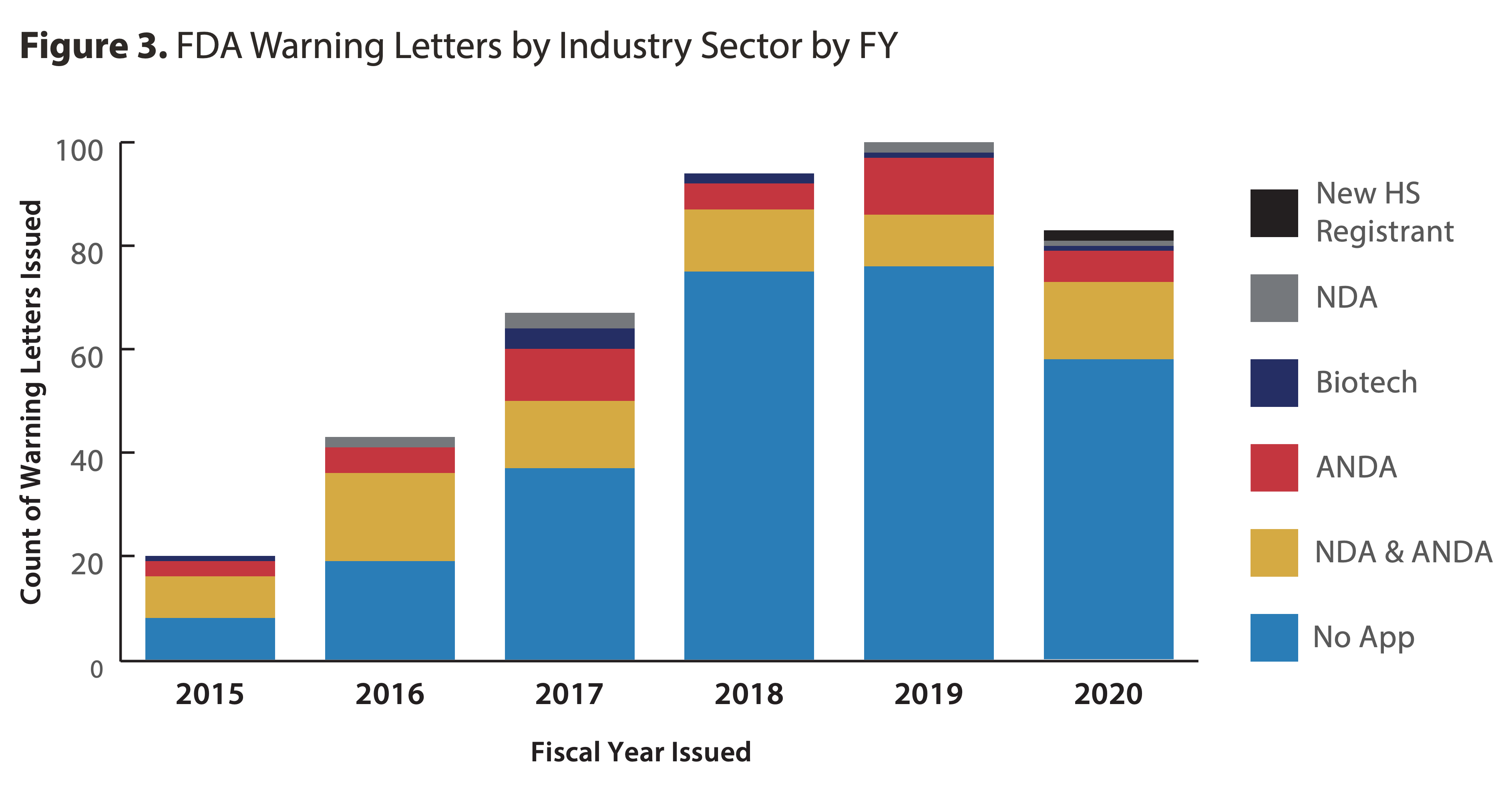 Source: fda.gov
Source: fda.gov
- “As in past years, the majority of Warning Letters in FY2020 were issued to sites with non-application products (69%), and especially those that manufacture finished dosage form (FDF), non-sterile, non-application products (41% of all Warning Letters).”
- “Import Alerts doubled to 128 in FY2020. Latin America had the most sites on Import Alert for the first time in FY2020, due to an unprecedented number of new hand sanitizer registrants from Mexico that failed to meet quality standards."
- Since FY2019, there has been a small decrease (0.10) in the mean Site Inspection Score (SIS) of the entire inventory of sites (7.3). FDA’s SIS, a scale of 1 to 10, is used as a proxy for compliance with CGMP regulations. The SIS is based on the classification of FDA drug quality assurance inspections conducted over the prior ten years, including inspections classified under the MRA program, which allows some global regulators to recognize reports from their counterparts’ inspections.
- “Since FY2019, there was a small decrease (0.10) in the mean SIS of the entire inventory of sites (7.3).”
- “For FY2016–FY2020, three defect categories account for 60% of all defects reported: Product Quality Questioned, Device Issues, and Packaging Issues.”
- In FY2020, the number of recall events increased for the second consecutive year.
- “The most recalled products by United States Pharmacopeia Therapeutic Category USPTC reflect the major quality issues over the past five years.”
- “The most substantial increases in the number of recalls by industry sector in FY2020 were in the No Application and NDA & ANDA (i.e., sites manufacturing for both application types) sectors.” (See full report for additional details.)
- “The average SIS of sites reporting at least one recall continues to be below the overall average SIS, highlighting the relationship between recalls and CGMP compliance.”
- “Each major recall over the last five fiscal years was associated with microbial or chemical contamination/impurities; a focus area for the industry to improve quality.”
- CDER will continue to seek to minimize long-standing problems such as drug shortages due to quality issues through proactive efforts including the New Inspection Protocol Project (NIPP) and Quality Management Maturity. The NIPP “is aimed at using standardized electronic inspection protocols to collect data in a structured manner. The protocols promote consistent and comprehensive coverage of critical areas of drug manufacturing and provide structured, data-rich reports.
- “The protocols include questions related to quality culture observed in facilities.”
- “In the future, FDA will have the ability to better understand how certain variables (e.g., location of the establishment, type of establishment) affect quality. As more data are collected through NIPP, these types of insights can inform future inspections, identify policy/outreach opportunities, and influence application-related decision making."
Pre-Pandemic FDA Inspection and Enforcement Trends (FY2019)
A report published by the ECA Academy looked back from October 2018 to September 2019 to review the FDA warning letter trends among pharmaceutical manufacturers.
Here, we’ve summarized this report’s findings, as well as those detailed in another comprehensive research report published on Pharmaceutical Online, which examined drug inspection observational trends by analyzing publicly available data from fda.gov.
Where applicable, we’ve provided links to relevant resources and next steps for those looking for guidance and assistance in mitigating trending risks.
1. Fiscal year 2019 saw the most warning letters issued to makers of finished medicinal products in recent history.
The last 12 months saw a total of 81 warning letters issued to finished product manufacturers worldwide, the most since the fiscal year 2015.
As ECA points out, this is in contrast to the number of warning letters sent to API manufacturers, which has fallen since peaking in 2017.
2. The majority of pharmaceutical warning letters were issued to US companies.
Over half (46 to be exact) of the 81 warning letters were issued to companies located in the United States. A detailed list of each region is below:
- United States: 46
- India: 12
- China: 10
- South Korea: 5
- Singapore: 2
- Costa Rica: 1
- France: 1
- Canada: 1
- Spain: 1
- Taiwan: 1
- Turkey: 1
3. The most frequently-cited GMP violations in warning letters concern basic requirements.
The specific issues contained within these recent warning letters reveal a continuation of a trend that’s been running for years: lapses in meeting basic GMP requirements.
The responsibilities of the quality control unit laid out in 21 CFR 211.22 were the top most-cited issue, followed by written procedures; deviations laid out in 21 CFR 211.100.
Here’s a more detailed breakdown of the top six pharmaceutical citations in the fiscal year 2019:
- 21 CFR 211.22 - 41 Warning Letters - Responsibilities of the quality control unit
- 21 CFR 211.100 - 31 Warning Letters - Written procedures; deviations
- 21 CFR 211.165 - 29 Warning Letters - Testing and release for distribution
- 21 CFR 211.192 - 27 Warning Letters - Production record review
- 21 CFR 211.166 - 22 Warning Letters - Stability Testing
- 21 CFR 211.84 - 22 Warning Letters - Testing and approval or rejection of components, drug product containers, and closures
Dive Deeper: Pharmaceutical Quality Assurance: FDA's Quality Unit Expectations
4. The number of inspection observations for drugmakers has increased.
As analyzed and compiled by Barbara Unger in an impressively researched column for Pharmaceutical Online, the frequency of Forms 483 issued to pharmaceutical companies has continued its steady rise over the past few years.
In the fiscal year 2019, there were a total of 779 Forms 483 issued for drug inspections compared to 716 in 2018, 694 in 2017, and 691 in 2016.
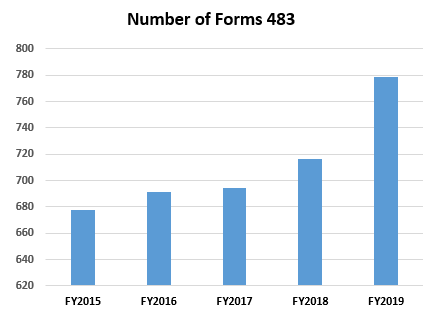
Source: FDA FY2019 Drug Inspection Observations And Trends, Barbara Unger, Pharmaceutical Online
Note that for this and the following sections covering inspection observation trends, the data presented adheres to the analysis methodology and limitations described in the introduction of the column referenced.
5. Six §211 citations increased dramatically between fiscal years 2018 and 2019.
Also noted in the column, “six regulations saw a significant increase in the frequency with which they were cited between 2018 and 2019. This includes:
- §211.100(a) Production and process controls shall be supported by written procedures
- §211.188 Master production and control records
- §211.113(b) Control of microbiological contamination
- §211.25(a) Personnel qualifications
- §211.165(a) Appropriate lab tests shall be used to determine conformance to specifications
- §211.67(b) Equipment cleaning and maintenance”
6. §211.22(d) was the most frequently cited compliance deficiency throughout fiscal year 2019.
Again, given the caveat of the limited public data available (the FDA’s data includes only Forms 483 issued through its electronic system and omits API manufacturers), some notable findings emerge regarding specific §211 citations.
The most frequent deficiency pertained to §211.22(d) Procedures applicable to the quality unit shall be in writing and shall be followed, which moved from the second most frequent citation to the first from the previous year.
The following list offers the top four most frequent citations throughout the last fiscal year.
- §211.22(d) Procedures applicable to the quality unit shall be in writing and shall be followed
- §211.192 Investigations of discrepancies
- §211.42(c) Facilities shall include defined areas of sufficient size
- §211.160(b) Laboratory controls shall include the establishment of scientifically sound and appropriate specifications, standards, sampling plans, and test procedure
The chart labeled “Table 1” in the column referenced offers a visualized breakdown of trending citations over the years. Access the full article here or open the chart (.png image file) in a separate window here.
7. OTC drugmakers were subject to greater scrutiny from regulators. Many issues point to basic GMP deficiencies.
As we explored in another article, 2019 saw the FDA put a greater compliance focus on over-the-counter (OTC) drugs and other health product manufacturers. In a July 2019 column for Pharmaceutical Online, we analyzed these OTC-specific compliance trends, pulling out the following common issues appearing in warning letters.
We’ve summarized the common themes of noncompliance below, but full details can be found in our article here.
- Documented Adherence to Relevant Regulations and Standards — Many recent warning letters to OTC manufacturers highlight fundamental issues related to the understanding and practical implementation of the 21 CFR 200 series, as well as relevant USP standards that address specific types of products and the systems used to produce them.
- Establishment and Suitability of Analytical and Microbial Testing and Validation Methods — Inadequate method development (method suitability and validation in particular) is another common theme in recent warning letters issued to OTC drug manufacturers. These critical processes are complex, often involving wide specifications, broad parameters, and the inherent variation that comes from working with living organisms.
- Nonconformance Management — Another recent trend afflicting OTC drugmakers mirrors a broader and well-documented trend throughout the drug and device space: inadequate nonconformance management. As demonstrated by a large number of citations issued specifically for “inadequate, incomplete, and undocumented investigations,” these warning letters offer evidence of a long-standing perception that an outsized focus is placed on immediate nonconformance correction rather than on thoroughly investigating and executing corrective and preventive actions following a comprehensive root cause analysis.
- Roles, Responsibilities, and Authority of the Quality Unit — The internal quality unit (QU) has been the target of many recent warning letters to OTC drug and health product manufacturers as an underlying cause of product quality and GMP compliance problems. Numerous firms have been cited for having an inadequate QU. In the most egregious examples, firms lacked this designated team entirely. More often, however, regulators have cited firms for a lack of written procedures that govern the responsibilities and functions of this group. 21 CFR Part 211 is clear about the need to establish a “quality control unit” with the documented responsibility and authority to make critical decisions.
Dive Deeper: 4 Trends In Recent FDA Warning Letters To OTC Drugmakers — And How To Avoid Them
Based on a growing number of relevant warning letters, as well as analyses of enforcement trend data and public statements made by the FDA, it’s clear that a renewed focus has been placed on evaluating manufacturers of OTC drug and health products in key areas of GMP.
These areas of enforcement focus include ineffective quality units, poor testing of incoming materials and components (i.e., relying on a supplier’s certificate of analysis), poor product testing, poor analytical and microbial testing and validation methodology (including method suitability), and inadequate nonconformance management.
Major Takeaways from the Report on the State of Pharmaceutical Quality: Fiscal Year 2019
Here are the major takeaways for inspections and compliance, specifically back from FDA's Report on the State of Pharmaceutical Quality: Fiscal Year 2019 :
- FDA investigators performed about 7% fewer drug quality surveillance inspections in FY2019 than they did in FY2018.
- The agency reviewed 109 drug quality inspections carried out by EU authorities under its good manufacturing practice (GMP) inspection mutual recognition agreement (MRA).
- 58% of inspections were conducted in facilities located outside the US.
- Based on its 10-point inspection score, the overall average for inspections in FY2019 was 7.4. (US- and EU-based sites scored averages that were slightly higher: 7.7 and 7.6, respectively.)
- Based on its 10-point inspection score, homeopathic products and sterile over-the-counter (OTC) products had the lowest average scores at 6.5 and 6.2, respectively. For more on recent chronic quality and compliance issues among OTC and health product manufacturers, read our deep dive.
- Three categories of inspection findings accounted for more than half of all observations:
- Records and Reports — CFR 21 Subpart J (23.9%)
- Laboratory Controls — CFR 21 Subpart I (19.3%)
- Equipment — CFR 21 Subpart D (14.8%)
- The most commonly cited CFR subparts were:
- 21 CFR 211.192, production record review (8%)
- 21 CFR 211.22, responsibilities of the quality unit (8%)
- 21 CFR 211.160, general requirements/scientifically sound laboratory controls (5%).
Takeaways, Resources, and Next Steps
Publicly available warning letters and inspection observation data provide powerful resources for understanding areas of regulatory focus and a benchmark for evaluating potential vulnerabilities within the quality system and beyond.
Download our relevant resources below for practical insight into mitigating many of the trending issues throughout the life sciences.
Relevant Resources

The OTC Drug Manufacturer's Guide to CGMP Compliance & Quality Management


Gap Analysis Remediation: A Guide to Resourcing & Implementation


The Complete Guide to Compliance Remediation Projects

Get Expert Quality & Compliance Assistance Now
In many of its warning letters, the FDA has “strongly recommended” engaging a third-party consultant qualified in the relevant regulations to assist in meeting CGMP requirements.
While we help many companies resolve Forms 483 and warning letters, we also help prevent them from being issued in the first place. At The FDA Group, we plan and conduct effective internal quality audits to ensure your quality system is completely aligned with all documentation and operations — the critical part of any internal audit.
More broadly, we assess your current systems and make the necessary improvements to ensure regulatory expectations are met across all functions of your organization. Read our case studies to learn how we've helped firms meet a variety of challenging life science goals.
In addition to assisting in a project-based consulting capacity, we help life science organizations fill key roles on their team through contracted and direct hire engagements. Explore our service areas to learn how we can locate, secure, and manage top industry talent, and/or work in a project-based capacity to evaluate, correct, and maintain your quality system.
![]() Need assistance now or planning for the future?
Need assistance now or planning for the future?
Get in touch with us today and get the conversation started