
The FDA’s recent and ongoing focus on over-the-counter (OTC) drugs and other health products intensified with the agency issuing warning letters to four companies with very similar citations.
The agency referenced these findings to point out its industry-wide concern for violations it said puts consumers at significant risk. These include insufficient quality controls for potentially hazardous products and skipped safety tests that have led to potentially harmful contamination in multiple products.
While these latest crackdowns target producers of homeopathic products specifically, (an area which began attracting greater attention from regulators back in 2015) the broader OTC manufacturing field has come under greater oversight, as evident by recent inspection and enforcement trends.
Analysts note significant increases in citations that send a very clear message to smaller firms and OTC manufacturers: Make sure you’ve established adequate controls for batch record development and execution and comply with CGMPs across the board.
This guide offers a clear and simple breakdown of the issues FDA has revealed (and are likely targeting during inspections), what those regulatory expectations are, and what OTC manufacturers should do to audit their manufacturing processes, reveal compliance gaps, and remediate them as soon as possible.

The OTC Drug Manufacturer's Guide to CGMP Compliance & Quality Management
Grab the free guide.

FDA Inspection & Enforcement Trends Reveal Fundamental CGMP Deficiencies in OTC Drug Manufacturing
Recent inspection and enforcement data reveal specific areas regulators are finding deficiencies in. This provides a useful roadmap for manufacturers to prioritize CGMP auditing and any necessary quality system implementation or remediation efforts.
A few relevant statistics make these priorities clear. (Note that the following analysis summaries don’t necessarily represent the FDA’s complete collection of inspectional observations, only those for which data is accessible for such analysis.)
- In FY2017, FDA inspectional data showed that the agency increasingly focused on over-the-counter (OTC) drug manufacturers, and many of these inspections cite either lack of, or incomplete, batch records. (Pharmaceutical Online)
- In FY2017, there was a dramatic increase in §211.188 citations (Master production and control records), specifically. The number of citations more than doubled from 100 in FY2016 to 208 in FY2017. (Pharmaceutical Online)
- In FY2018, the OTC non-prescription drug group was deemed a breakaway category in regards to the types of manufacturers that received FDA warning letters. Over half (53%) of the warning letters to drug product manufacturers were issued to OTC manufacturers. (Pharmaceutical Online)
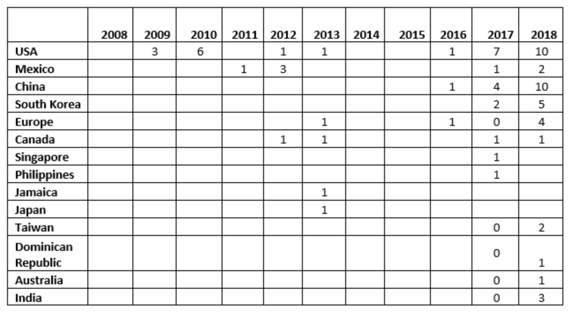
- Between CY2017 and CY2018, the number of warning letters issued to OTC manufacturers more than doubled, from 17 to 39. More countries, 10 vs. seven, were associated with these warning letters in CY2018. (Pharmaceutical Online)
Warning Letters Issued to OTC Manufacturers by Country, by CY
 (
(
Additionally, the most recent warning letters (most being issued in 2019) further underscore FDA's focus on inspection and enforcement of OTC/health product manufacturers in key areas, as shown in a sampling of examples below.
Tec Laboratories
- Your firm failed to conduct appropriate laboratory testing, as necessary, of each batch of drug product required to be free of objectionable microorganisms (21 CFR 211.165(b)).
- Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192).
Red Mountain, Inc.
- Your firm failed to establish a quality control unit with the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging materials, labeling, and drug products (21 CFR 211.22(a)).
- Your firm’s quality control unit failed to approve or reject all procedures or specifications impacting on the identity, strength, quality, and purity of the drug product (21 CFR 211.22(c)).
Read the full FDA Warning Letter →
King Bio, Inc.
- “Your firm failed to establish an adequate quality control unit with the responsibility and authority to approve or reject all components, drug product containers, closures, in-process materials, packaging materials, labeling, and drug products (21 CFR 211.22(a)).
- Your firm failed to establish adequate written procedures for production and process control designed to assure that the drug products you manufacture have the identity, strength, quality, and purity they purport or are represented to possess (21 CFR 211.100(a)).
- Your firm failed to use equipment in the manufacture, processing, packing, or holding of drug products that is of appropriate design, adequate size, and suitably located to facilitate operations for its intended use and for its cleaning and maintenance (21 CFR 211.63).
Read the full FDA Warning Letter →
Beauty Manufacturing Solutions Corp.
- Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192).
- Your firm failed to establish laboratory controls that include scientifically sound and appropriate specifications, standards, sampling plans, and test procedures designed to assure that components, drug product containers, closures, in-process materials, labeling, and drug products conform to appropriate standards of identity, strength, quality, and purity (21 CFR 211.160(b)).
Read the full FDA Warning Letter →
Pure Source, LLC
- Your firm failed to thoroughly investigate any unexplained discrepancy or failure of a batch or any of its components to meet any of its specifications, whether or not the batch has already been distributed (21 CFR 211.192).
- Your firm failed to establish and document the accuracy, sensitivity, specificity, and reproducibility of its test methods (21 CFR 211.165(e)).
As industry analysts and the recent enforcement actions have noted, the takeaway for OTC manufacturers is clear:
The FDA is finding that many OTC manufacturers are failing in the fundamentals of CGMP. Key areas of deficiency include ineffective quality units, poor testing of incoming materials and components (i.e. not relying on a supplier’s certificate of analysis), poor product testing, poor analytical and microbial testing and validation methodology (including method suitability), inadequate nonconformance/CAPA management, and failure to have real-time data for expiry labeling.
In response, OTC and health product manufacturers should carefully assess their own state of CGMP compliance with special attention given to priority areas and perform the necessary remediation to bring their organization into full compliance with complete documentation each step of the way.
A Quality Systems Approach to CGMP OTC Manufacturing Compliance
OTC drug manufacturers, labelers, packagers and holders must comply with current good manufacturing practices — a system of controls intended to help to prevent exactly the kinds of issues investigators are finding at OTC production facilities: contamination, mix-ups, deviations, failures, and errors.
The quality systems approach to CGMP compliance offers a methodology specifically designed to help firms reveal current compliance gaps and create a manageable, controlled system for meeting the standards set for manufacturing and testing in areas such as raw materials, quality assurance, record-keeping throughout the manufacturing process, standards for cleanliness and safety, qualifications of manufacturing personnel, product testing, production and process controls, and warehousing and distribution.
FDA’s guidance document on this approach describes a full quality systems model, pointing out the approach’s consistency with CGMP regulatory requirements for manufacturing human and veterinary drugs as well as biological drug products.
It also describes how manufacturers implementing quality systems can be in full compliance with the regulations coming under intense scrutiny: 21 CFR parts 210 and 211. It reviews CGMPs, the concepts of modern quality systems and how such a system can be implemented.
"CGMP Consultant Recommended"
When reviewing the warning letters listed above, a common FDA recommendation emerges: "CGMP Consultant Recommended."
An excerpt from one warning letter offers a look at where within their systems regulators recommend firms get expert assistance to both audit and remediate these issues:
Whether implementing a quality systems approach or simply improving upon a current quality assurance system, the path to compliance remediation is often complex. It requires cross-functional cooperation, strong project management, and the experience necessary to anticipate pitfalls and implement best practices.
Leaving critical auditing tasks to an entirely internal team often invites the natural, unseen biases that prevent objective assessment. These projects can also suffer from internal myopia when regimented routines and methods prevent firms from taking advantage of novel opportunities for improvement.
For these (and many other) reasons, compliance assessment and remediation is often best managed with the help of a third-party quality consultant — an objective outsider who has seen and solved a variety of issues with similar firms and can utilize this experience to bring a firm into compliance and make sustainable process improvements along the way.
In an increasingly complex regulatory environment, experienced quality and compliance consultants have become invaluable guides to help firms orient and navigate where to go and how best to get there. This is especially true in situations where greater regulatory oversight requires firms act quickly.
Get Expert CGMP Auditing & Remediation Assistance Now
Ensuring compliance with CGMPs is a challenge we help pharmaceutical firms of all kinds overcome through comprehensive auditing and remediation projects led by experienced quality and compliance experts backed by a total quality guarantee.
Contact us today to learn how we can help you understand your quality and compliance needs, audit and remediate your current systems, and protect your reputation for producing safe, high-quality products.
The OTC Drug Manufacturer's Guide to CGMP Compliance & Quality Management
Grab the free guide.