
“Insufficient corrective and preventive action procedures” has consistently topped the list of most common FDA inspectional observations within the medical device industry since the fiscal year 2010.
Its prevalence as the top problem year after year makes it clear that many device companies have issues, both known and unknown, within their CAPA programs.
While the immediate compliance threats are obvious, less so are those that leave companies vulnerable to serious quality system issues that can grow and metastasize under the radar of their quality management system, putting both patients and their businesses at risk.
This guide tackles CAPA from a slightly different perspective than most are used to seeing in industry publications and seminars.
We’ve gathered insights from the quality and compliance experts who have seen it all and fixed it all firsthand. In addition to exploring the solutions that have proven to be effective in the field, we’ll also examine one, if not the, most pivotal component of CAPA: root cause analysis.
Table of Contents
Use the links below to jump around this guide.
- CAPA 101: The Basics
- What the FDA Expects From Your CAPA Process
- Root Cause Analysis: A Practical Perspective
- An Informed Approach to Addressing and Preventing "Human Error"
- Determining When CAPA is Appropriate
- How CAPA Interfaces With Your Quality Management System
- The Anatomy of an Effective and Compliant CAPA Form
- An Adaptable CAPA Model You Can Use
- Common CAPA Problems to Avoid
- The Value an Expert Third-Party Consultant Can Bring to Your CAPA Program
- Get Expert Assistance
Since this guide is comprehensive, we suggest downloading our PDF version, which you can access, save, and use below.
 TAKE THIS GUIDE WITH YOU
TAKE THIS GUIDE WITH YOU
The Guide to CAPA & Root Cause Analysis in FDA-Regulated Industries
Download the full white paper version of this guide below. It offers everything we cover here and more in a handy PDF you can use to assess and enhance your CAPA program.

Meet the Contributors

.png?width=144&name=circle-cropped%20(2).png)
Watch our free CAPA webinar
CAPA 101: The Basics
What is CAPA?
According to the U.S. Food and Drug Administration (FDA), corrective and preventive action aims to collect information, analyze information, identify and investigate product and quality problems, and take appropriate and effective corrective and/or preventive action to prevent recurrence.
Verifying or validating corrective and preventive actions, communicating corrective and preventive action activities to responsible people, providing relevant information for management review, and documenting these activities are essential in dealing effectively with product and quality problems, preventing their recurrence, and preventing or minimizing device failures.
Corrective and Preventive Action (CAPA) was first formally introduced by the U.S. Food and Drug Administration (FDA) in 2006 as a component of the Quality Systems Guidance. This guidance would go on to form the basis of the ICH Guideline Q10. Since then, it’s found its way into the EU GMP Guide, laying out the CAPA process within the pharmaceutical space.
For medical device companies, CAPA is addressed in ISO 13485, which, unlike Q10, divides the concept into its two concepts: “Corrective measures” (addressed in Chapter 8.5.2) and “Preventive measures” (addressed in Chapter 8.5.3). Despite separating these processes, both must be documented and evaluated to demonstrate improvement and preventive action, making CAPA the practical process by which both are united. With this in mind, we’ll treat it as one concept here.
What is CAPA used for?
A CAPA procedure addresses deviations or problems that have already occurred and puts measures in place to avoid future deviations or problems. This entire process of analyzing errors, deviations, and their effects can—and should—be carried out as a component of broader risk assessment rooted in a well-defined and documented risk management program.
CAPA-triggering deviations can originate from a variety of sources within a quality management system, such as internal audits, customer feedback, or in the most serious cases, safety or security-related incidents that result in faulty products due to inadequate controls.
No matter why or where CAPA is initiated, it should always begin by identifying and taking immediate actions to stabilize the situation and limit its further effects. This is where an honest and accurate assessment of its severity is absolutely crucial. In the worst-case scenarios, this could mean an immediate halt to the production and distribution of all impacted products.
What does a typical CAPA process look like?
In most cases, a CAPA is executed much like a typical PDCA (Plan-Do-Check-Act) cycle:
- Begin with a detailed problem description by accurately evaluating and documenting it. Similar to the planning stage of the PDCA cycle, all CAPAs should start by describing the problem in detail, ensuring everything is documented accurately.
- Perform a root cause analysis. Once the problem is clearly defined, determine its root cause by performing a thorough root cause analysis.
- Implement, verify, validate, and document necessary corrective and preventive measures. This change should seek to remedy the root cause of the problem and ensure no further problems will occur. This could be a change in the production process, a training improvement, or another transformative measure.
- Check for effectiveness and evaluate success. Finally, the effectiveness and success of the measures implemented is checked and evaluated accordingly. Like with the “Act” phase of PDCA, this justifies whether and why the CAPA process was successful. If the CAPA process was not or only partially successful, another CAPA process must be initiated. A successful CAPA process may result in a re-evaluation or addition to the risk analysis.
The diagram below offers a simplified look at the general steps of CAPA. An adaptable model for CAPA is offered later in the paper.
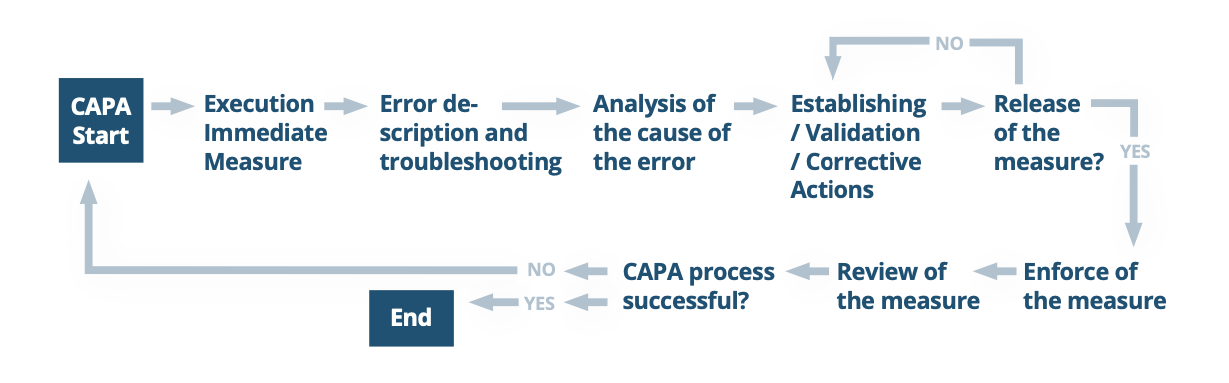
What the FDA Expects From Your CAPA Process
Given the numerous inspectional observations citing insufficient established CAPA procedures, it’s worth revisiting what regulators expect to see from your process.
In a 2014 presentation, FDA’s Joseph Tartal described the basics of effective corrective and preventive action—a resource every company should use to evaluate against their own processes. Among many important points, one stands out as particularly useful when ensuring your procedure meets regulators’ expectations:
“Manufacturers should consider that their corrective action and preventive action documentation can demonstrate to FDA that the manufacturer’s quality system is effective and enables the manufacturer to identify problems quickly and implement effective corrective and preventive actions (or not).” — Brian Dense
In addition to helping manufacturers meet the broad expectations for effective CAPA, FDA also makes public its own inspectional guide, which lays out the specific objectives for investigators when evaluating a device company’s CAPA system and related documentation.
We’ve summarized its main points below.
This should serve as your ultimate preparedness checklist when evaluating your CAPA processes for compliance with FDA regulations.
-
Verify that CAPA system procedure(s) that address the requirements of the quality system regulation have been defined and documented.
-
Determine if appropriate sources of product and quality problems have been identified. Confirm that data from these sources are analyzed to identify existing product and quality problems that may require corrective action.
-
Determine if sources of product and quality information that may show unfavorable trends have been identified. Confirm that data from these sources are analyzed to identify potential product and quality problems that may require preventive action.
-
Challenge the quality data information system. Verify that the data received by the CAPA system are complete, accurate, and timely.
-
Verify that appropriate statistical methods are employed (where necessary) to detect recurring quality problems. Determine if the results of analyses are compared across different data sources to identify and develop the extent of product and quality problems.
-
Determine if failure investigation procedures are followed. Determine if the degree to which a quality problem or nonconforming product is investigated is commensurate with the significance and risk of the nonconformity. Determine if failure investigations are conducted to determine the root cause (where possible). Verify that there is control for preventing the distribution of nonconforming product.
-
Determine if appropriate actions have been taken for significant product and quality problems identified from data sources.
-
Determine if corrective and preventive actions were effective and verified or validated prior to implementation. Confirm that corrective and preventive actions do not adversely affect the finished device.
-
Verify that corrective and preventive actions for product and quality problems were implemented and documented.
-
Determine if information regarding nonconforming product and quality problems and corrective and preventive actions has been properly disseminated, including dissemination for management review.
⚠️ A few action items to consider:
- Evaluate your current CAPA process on the criteria listed above.
- Highlight and remediate any gaps that exist between regulatory expectations and current processes.
- Follow up on all changes with the necessary documentation, training, or other actions needed to implement, support, and maintain those improvements.
- Note any gaps or improvements that require third-party assistance from a qualified CAPA professional and contact a firm to pair you with the appropriate resource.
Need expert assistance with any of these considerations? Get in touch with us to discuss the best way to close gaps, ensure compliance, and enhance your CAPA program.
Root Cause Analysis: A Practical Perspective
While much attention is given to the tools, techniques, and methodologies for “extinguishing the problem at the source,” much less attention is given to the fact that none of them will be effective if they aren’t implemented properly.
The factors for proper use boil down to two fundamentals every organization should immediately evaluate against and enhance if room for improvement exists:
-
Having the right team(s) in place to collect data and conduct the investigation in order to determine what factors should and shouldn’t be included in the analysis
-
Crystal clear communication with the proper measures in place to minimize bias and the role of inter-organizational politics in obstructing the free and open exchange of facts and ideas
Without these two fundamentals, even the best root cause analysis process will likely fail to properly identify and address the true root causes of the problems affecting your products and quality system.
Once a foundation is established on these two important pillars, turn your attention to the major challenges companies face in conducting root cause effectively.
- Challenge #1: Devoting too little time to investigating and determining the root cause of an issue
- Challenge #2: Simply restating the problem statement as the root cause of the problem
- Challenge #3: Not having a reliable set of tools and methods for carrying out an effective investigation
- Challenge #4: Failing to reference the tools you do use in your CAPA procedure, thereby opening the door to regulatory scrutiny
Since each company faces its own set of challenges, prescribing a universal set of solutions is impossible. A solution for one organization can end up introducing more problems when applied the same way in another.
It’s important to reiterate that this guide is intended to reveal the realm of possible solutions and improvements available to you—not compel you to use one or another. Given this frame, let’s explore a topic that’s familiar to many, but provides the critical foundation from which action is taken: root cause analysis.
“In many organization, politics are allowed to influence the CAPAs simply because there are too many people involved in the approval process—approval to actually open a CAPA, or approval to finish one. Very often there are too many people, or the wrong people, in the approval process. I’ve seen real problems get swept under the rug because somebody won’t approve the effort to go forward with it. I’ve also seen good solutions get shot down on real problems, because somebody didn’t want to spend the money or the time. I would be cautious about adding too many levels and too many people to the process.” — Brian Dense
A Brief Overview of Root Cause Analysis Methodologies
There are a number of reliable methodologies for analyzing root cause, however, not all are equally effective in every scenario. Applying the same methodology to every investigation can fail to go far or wide enough into the problem, undermining the entire effort.
While each methodology deserves to be thoroughly understood by those putting them to use, we’ve summarized the key takeaways for three of the most common models to help you choose the right one for a given scenario.
Fault Tree Analysis (FTA)
This deductive procedure is used to determine the various combinations of hardware and software failures and human errors that could cause undesired events (referred to as top events) at the system level.
The generic example below was developed for sixsigmastudyguide.com's FTA explainer:
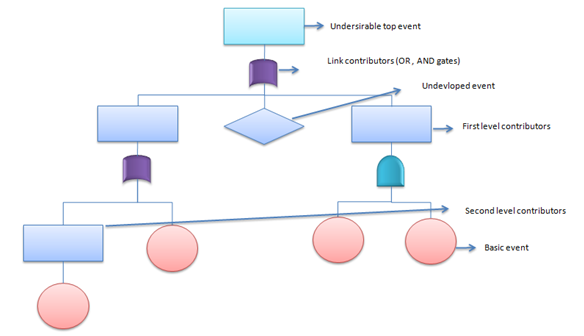
- Advantages: FTA values the judgment of experts from many disciplines and provides a common perspective on a given problem.
- Disadvantages: FTA relies on multiple expert opinions and judgments at various stages, making it prone to inaccuracy and bias. For larger systems, the quantitative analysis might be so complex, it requires computer algorithms to accomplish.
- Recommended Applications:
- Understanding the logic leading to a “top event” or undesired state
- Demonstrating compliance with system safety and reliability requirements
- Prioritizing multiple contributors which led to a top event or undesired state Recommended Applications
- Analyzing and forecasting system reliability, maintainability, and safety
Fishbone Diagram
Also called a “cause and effect” or “Ishikawa” diagram (among others), a fishbone diagram is a visual tool for looking at cause and effect. A problem or effect is displayed at the “head” or “mouth” of the fish, and possible contributing factors are listed on the “bones” under various cause categories. These models work best when the “head” of the fish contains a very detailed problem statement. This helps eliminate the scope creep of the team’s discussions. What happened? When? Where? These can help narrow the focus to solve the problem.
Here's an example pulled from tulip.co's explainer guide on the concept:
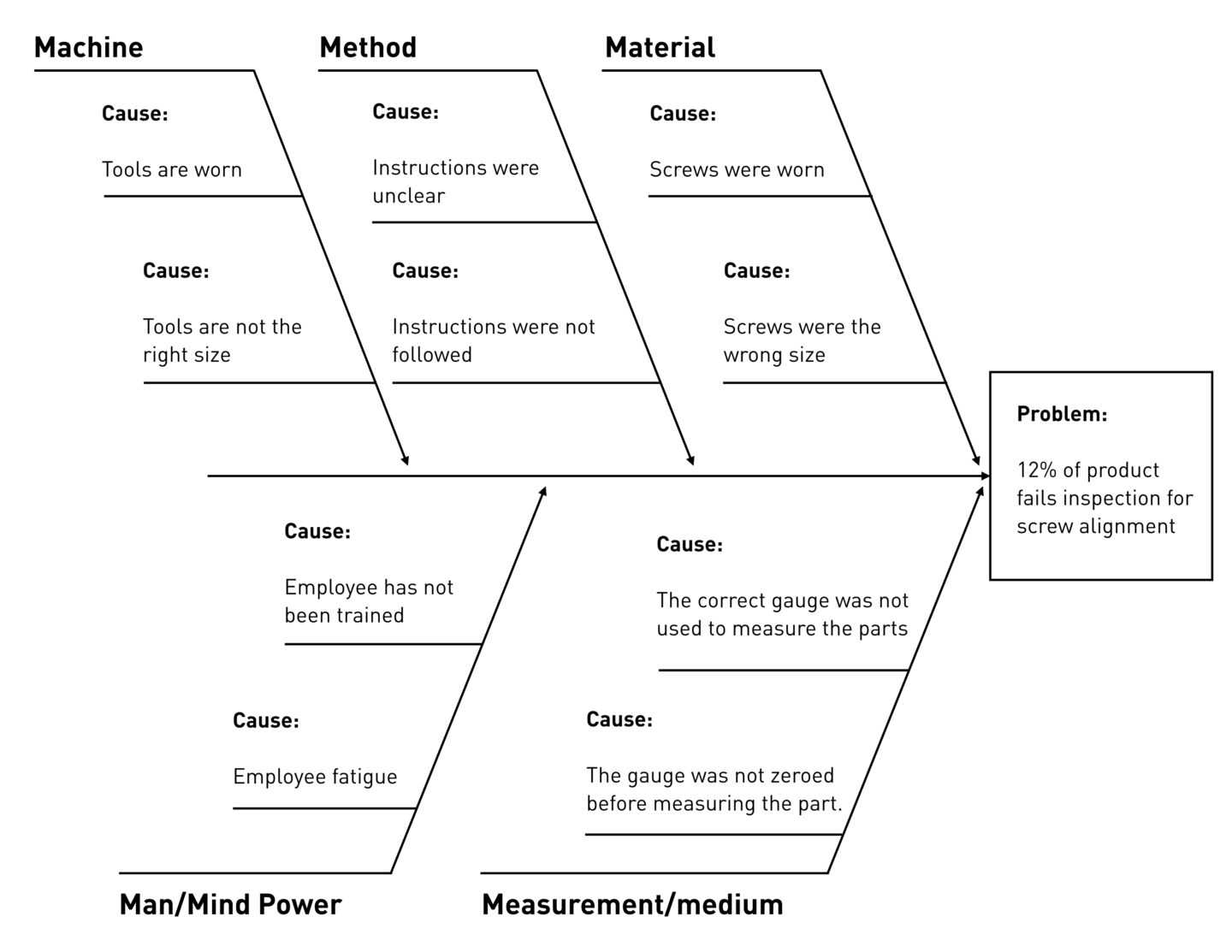
- Advantages: Fishbone diagrams are particularly useful for organizing potential causes, helping teams think through causes they might otherwise miss, and providing a living document that shows the status of all potential causes and whether they have been proved, disproved, or acted upon.
- Disadvantages: Fishbone diagrams are by nature a divergent approach to problem-solving, so they make it possible for teams to expend a lot of energy speculating about a potential cause that may have no effect on the problem.
- Recommended Applications:
- Directing a team to focus on identifying all possible categories and consider alternative causes
- Refocusing a team on the causes of a problem rather than the symptoms
- Improving product design
- Preventing quality defects
- Identifying potential factors causing an overall effect
5 Whys
The 5 Whys is arguably the simplest technique for root cause analysis. It can be very effective when answers come from people with hands-on experience in examining the process. It is remarkably simple: when a problem occurs, you drill down to its root cause by asking “why?” five (or more) times. Then, when a countermeasure becomes apparent, you follow it through to prevent the issue from recurring.
- Advantages: This is essentially a simpler form of a fault tree analysis, making it a straightforward process when investigating specific accidents instead of chronic problems.
- Disadvantages: Results can be non-reproducible and inconsistent. For instance, two teams analyzing the same issue may reach a different solution. It also leaves the door open to stopping short of reaching the true root cause.
- Recommended Applications:
- Resolving simple or moderately difficult problems
- Resolving issues involving human factors
- Resolving issues where statistical analysis is not needed or possible
The Importance of Cross-Functionality When Conducting Root Cause Analysis
Today, it’s common for organizations to place a high value on taking personal responsibility for quick problem resolution. While this value is rooted in good intention, placing the focus at least in part on speed must be done with extreme caution to ensure root causes are fully identified and resolved.
Speed, in this case, is a risky substitute for thoroughness. When this risk is ignored, the incentive to kick the can down the road to get it cleared and closed as fast as possible begins obstructing your ability to conduct a thorough investigation and analysis. Rather than resolving problems, they’re simply moved from one function to another in a dangerous game of hot potato.
Processes that give rise to problems are rarely localized to a single department or function. In many cases, the more complex a process is, the more functions it crosses. To truly resolve problems at their root—quickly and completely—a cross-functional group consisting of stakeholders from all inputs, work tasks, and outputs involved must be established to solve the problem from every angle.
To be reliably successful, root cause investigation and analysis must start by defining the process in which an issue (or issues) have arisen from one end to the other—evaluating all inputs, work done, and outputs being handed off to the next process. To do this effectively, knowledgeable representatives and owners from all functional areas must work together, including engineering, sales, quality, regulatory, etc..
Cross-functionality is especially important given that the point at which a problem is detected is rarely where the root cause truly lies. The further upstream you need to travel to find it, the more you can expect to rely on the knowledge of other functions you’re led to when tracing the principal cause. This is where communication and cooperation between functions become critical. Without a cross-functional approach, assumptions made by one function conveniently replace the informed knowledge of another—leading to dangerous gaps in understanding how other steps in the process are impacted.
Creating a cross-functional team may slow down the process and ask more of those involved, but this added investment is often returned in the quality of the results it achieves.
⚠️ A few action items to consider:
- Evaluate the current methodologies and tools utilized when investigating the root cause.
- Determine whether current processes suffer from the challenges described and consider ways to overcome them.
- Determine how well current processes ensure the best methodologies and tools are selected from diverse options based on applicability to specific goals.
- Adjust processes as necessary to include currently unused methodologies and tools given staff are fully trained and confident in using them.
- Consider how prior problems could have been solved better if addressed with another methodology or tool and prepare to employ them if similar issues arise in the future.
- Evaluate your current root cause analysis process on its degree of cross-functionality.
- If needed, revise this process to incorporate more stakeholders from other functions.
- To accelerate problem resolution, establish the team’s assignment and timeline up-front.
An Informed Approach to Addressing and Preventing "Human Error"
Genuine human errors do happen, but they’re cited far more frequently than they should be. In truth, most problems that appear to be caused by human error— especially those that occur multiple times—are actually rooted in processes or systems that when left unchanged, will keep producing the problem despite the convenient band-aids often placed over them.
When human error is identified more frequently than it should be expected to happen, it signals to investigators that problems aren’t being investigated thoroughly enough, thereby shifting them into problem-hunting mode and opening your quality management system up to even greater scrutiny.
But what about the rare instances where one-time errors are made by otherwise well-trained personnel following well-defined processes? A moment of inattention due to a passing distraction can lead to serious problems. In these cases, the human error classification may be justified after a thorough investigation reveals nothing in every possible place there is to look. Again, this conclusion should never be jumped to as a convenient way to avoid the important work of problem-solving (as it often is). It should only be considered as a viable justification when every other possible cause has been exhaustively explored and eliminated.
Given this caveat, it’s helpful to arm yourself with a model for analyzing what might appear to be human errors in order to determine whether actions (or inactions) were deliberate or inadvertent. The outcome can help you determine whether or not human behavior really is to blame as well as where you might expect to find a problem elsewhere (such as inadequate training, poor SOPs, etc.) if the root cause appears to be less human than you initially thought upon further analysis.
Enter the Skills, Rules Knowledge/Generic Error Modeling System illustrated below. It’s relatively simple but offers a reliable way to tease out genuine human errors from other problems while pulling real human errors apart even further to reveal their motivating factors.
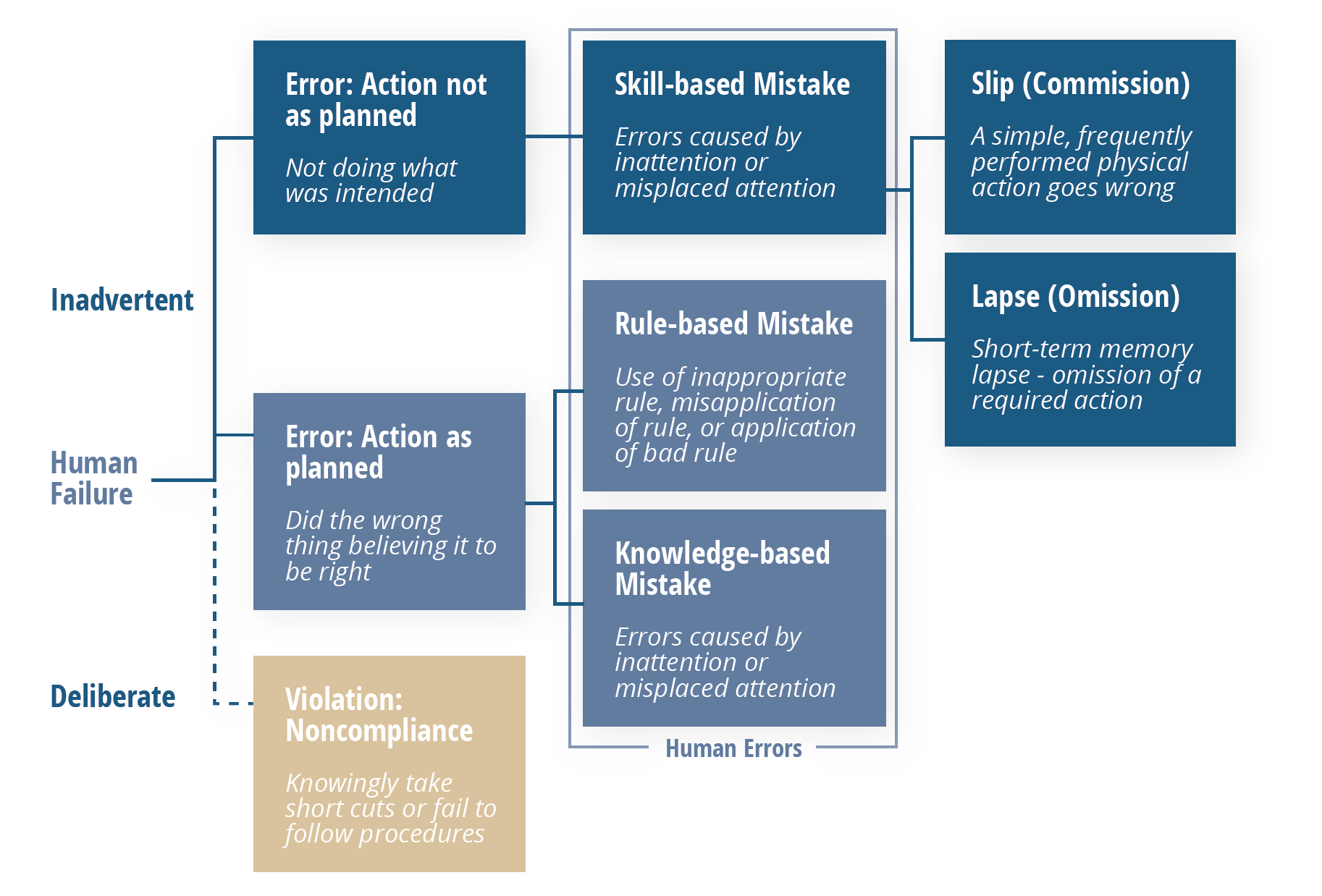
Following this model, errors that are shown to be “inadvertent” can be considered genuinely “human”, which then fall into one of three categories: skill-based, rule-based, or knowledge-based mistakes.
Those that are skill-based can be broken down one more time, into either a slip or a lapse. Both of these point to the same root cause: a lack of attention, which manifests itself in two different ways: momentary memory loss or a routine action that wasn’t performed.
Once more, it’s important to note that these types of errors should not be happening frequently. The vast majority of problems that appear to be a human error at first should lead you elsewhere upon further analysis.
Errors shown to fall into one of the two other categories should be viewed through a different lens. In these situations, processes—particularly related to training and oversight should be scrutinized as contributors as explained in the breakdown below.
Knowledge-based Errors
- Were they tasked with doing too many things at once?
- Is that department under-resourced?
- Was this individual’s supervisor aware that the person is multitasking beyond their limit?
Rule-based Errors
- Did they receive insufficient practice in training?
- Did practice actually reflect the operations they were performing on the floor?
- Does the procedure specify the details to the degree they need to be explained in order to be performed correctly?
Read our companion article from which this section is excerpted to learn two places to look for systemic human errors: An Informed Guide to Addressing and Preventing Human Error.
⚠️ A few action items to consider:
- Conduct an objective assessment of your internal problem-solving processes (ideally with the help of an experienced third party) and remediate accordingly.
- Replace metrics that establish problematic incentives with goals focused on long-term trending.
- Explore ways to improve problem-solving within your QMS to reduce backlogs while thoroughly investigating issues.
Determining When CAPA is Appropriate
Many companies suffer from either overusing or underusing their CAPA program— each presenting its own set of risks. Overusing CAPA typically results in a never-ending backlog of open projects, which prevents issues from being resolved in a timely manner, thereby allowing them to worsen while in queue. Underusing CAPA risks letting issues fly under the radar, which again enables them to grow and become harder and more expensive to fix when they’re finally detected.
It would be reckless to prescribe hard-and-fast rules for when or when not to open a CAPA, but for those struggling with over or under-use, some general advice may be useful for taking immediate initial steps to correct yourself and set the stage for gradual improvement.
- Ramp up or scale back your CAPA program to deal with systemic or potentially systemic issues first. While not every problem deserving of a CAPA should be “systemic” in nature, these types of problems are typically a great fit when addressed with CAPA.
- Ask four questions of the problem:
- Did the incident or event result in injury? If so, CAPA should obviously be mandatory.
- Has the issue, incident, or event occurred multiple times? If so, you likely have a system problem on your hands, which should definitely be handled through CAPA.
- Can the issue be managed effectively another way? “Effectively” is the keyword here. This question shouldn’t be used as an excuse to not open a CAPA if it’s necessary, but if other methods are worth considering, they should be explored.
- Does the issue appear to be critical following a risk assessment? High-risk issues like those deemed to be “critical” should be addressed through a formal CAPA.
-
Use creative trending techniques. Despite no hard-and-fast criteria for CAPA, quality system expert Brian Dense explains that trending data can be used to make informed decisions about the types of problems CAPA should be considered for while also reducing the amount of work involved.
“If companies did appropriate trending, they could actually combine and condense a lot of the work they’re doing in their CAPA program. You may have five different corrective action efforts going on at once, but you might have five problems that are each only slightly different. After investigating, you might discover they all have the same root cause. At that point, you should consider moving all of these CAPAs into one. Close the others, so that you’re not spending all this time worrying about a record. Have good procedures, with clear rationale. Give a good, solid justification for why you’re doing what you’re doing. Close the record, and put your focus on the best one to implement the corrective action. Trending helps companies reduce the amount of work they actually have to do within their CAPA programs.” — Brian Dense
⚠️ A few action items to consider:
- Consider establishing a system for recording internal CAPA trends in order to better use your program to address appropriate issues.
- Consider using general questions to better screen issues resulting in a CAPA.
How CAPA Interfaces With Your Quality Management System
Corrective and preventive actions should always be utilized when necessary throughout any area of your organization. Given this very important point, it’s also helpful to know which systems typically give rise to the problems CAPA is used to solve (and prevent). While again, these are by no means the only areas to expect issues to occur, it can help you prioritize regular monitoring and realize the true scope of problems by identifying their early indicators.
- Nonconforming Product: While one-time nonconformance issues aren’t necessarily worthy of a CAPA, systemic issues almost always are. Confident decisions here rely on your process for analyzing nonconformance data. This is one area where effective analysis can help you be proactive rather than reactive to addressing situations.
- Complaint Handling: Complaint handling processes are critical from FDA’s perspective. Investigating complaints requires a thorough process very similar in concept to a CAPA process. It should define the issue, determine root cause, and establish a plan to address and correct the situation. When this process is effective, CAPA should mainly be reserved for systemic issues. Again, this requires monitoring and analyzing to know when in fact complaints are being caused by an underlying systemic issue.
- Production & Process Controls: In most cases, product issues identified during the production process should be captured as nonconformances—the initial way issues with out-of-spec products are addressed. If the nonconformance is systemic, CAPA will likely be necessary. In other cases, you may identify issues with production-related processes. These require smart corrective and preventive action. While many of these can be addressed via your change-management process, again, CAPA should be considered when issues are revealed to be systemic.
- Supplier Management: Supplier issues that are beyond the scope of a nonconformance—again, mainly those shown to be systemic, should be dealt with either via internal CAPA or a supplier corrective action request (SCAR). Issues elevated and floated back to the supplier should be closely tracked and documented.
- Audits: While audits are typically seen as straining and time-consuming, they can be great opportunities for making improvements and discovering potential issues before they become problems. Once again, any systemic issues revealed during an audit should be prime candidates for a CAPA investigation.
The Anatomy of an Effective and Compliant CAPA Form
In addition to being fully compliant with ISO 13485:2016 and 21 CFR 820.100, your CAPA procedure must also be effective. This means it has to be clear and functional for its users, the majority of whom likely aren’t CAPA experts.
We've outlined the high-level components of a CAPA form below. Download our accompanying white paper for more detail into each.
- Date of initiation
- Cross-reference number
- CAPA source
- Description of CAPA issue
- Investigator assigned and target due date
- Investigation of Root Cause
- Containment of nonconforming product
- Correction(s)
- Corrective action plan/target deadline for implementation
- Preventive action plan/target deadline for implementation
- Corrective and preventive actions implemented
- Plan for Verification of Effectiveness
- Verification of Effectiveness
- Signature and Closure Date
An Adaptable CAPA Model You Can Use
While CAPA doesn’t lend itself to a universal model or set of procedural steps, the high-level steps presented below offer a dependable baseline from which to compare your current process, build the foundation of a new one, or adjust to improve its effectiveness in correcting and preventing problems.
1. Create and Submit a Request
Begin with a request outlining the possible causes and probable sources.
“Do you have a document that describes how to request a CAPA? You need to have a way for personnel to approach the quality group and provide the critical information needed to request this.” — Larry Stevens, RAC
2. Review the Request Appropriately
CAPA request review should be handled by a Quality Manager or Quality Review Board to determine if it’s warranted. Ideally, a QRB should meet regularly to review CAPA requests and other open quality matters. If rejected, the rationale must be documented in case the issue resurfaces. If initiated, it should be assigned a unique sequential ID number and moved forward.
“Requests need to be documented and reviewed within a certain timeframe. If it’s rejected, that rationale needs to be documents and placed where requests are filed. Make sure you can justify these decisions on a risk basis. If you do open a CAPA, it needs to be uniquely identified and given a CAPA owner—typically the person who requested it who takes responsibility for moving it forward.” — Larry Stevens, RAC
3. Finalize Sources and Build Your Team
Identify all personnel, processes, procedures, and functions that could be involved with the CAPA and document in full. Create a team responsible for conducting the investigation and creating the action plan. Ensure your team is cross-functional and inform them of their roles in the investigation.
“Create a cross-functional team that will meet on these matters. Any individual CAPA may have a team of anywhere from three to five (or more) people. The CAPA owner, then, will ensure the members of the team understand what they’re doing and hold them accountable to the action plan that was created, documenting the progress as it evolves.” — Larry Stevens, RAC
4. Identify Immediate Corrective (and Other) Actions
Detain nonconforming products or materials. Take any other immediate steps to correct glaring problems and document in full.
“CAPAs dealing with nonconforming products often require quarantining those products and getting them out as an immediate step. Other types of glaring problems might be an employee incorrectly assembling a piece of equipment. That needs to be fixed right away while analyzing the reasons why that problem happened.” — Larry Stevens, RAC
5. Investigate and Record Findings
The cross-functional team should conduct a thorough investigation. Very importantly, the degree of effort, resource investment, and documentation should correspond to the level of risk for the given problem.
“A good record of your investigation should be generated. And please tell your engineers, or whoever is writing this that they should not be writing it for other employees to read. It should be written for FDA or outside auditors to read. Put enough detail into it so it doesn’t raise more questions. A “lack of training” resulting in “additional training” only raises more questions. What additional training was done? Who did it? How was it documented? These reports should be very detailed and written so that someone reading it gets the full story. If you give that document to FDA and the details aren’t there, you just told them you didn’t do anything.” — Larry Stevens, RAC
6. Determine Root Cause
Select an appropriate analytical approach and use it to arrive at the root cause of the CAPA. Remember that effective action plans require accurate analysis, so this step is absolutely critical.
“As part of that analytical procedure, you need to identify the tools you’ll use for root cause determination and create a formal document that identifies the root cause. This is a critical step in defining your action plan.” — Larry Stevens, RAC
7. Develop, Execute, and Implement Your Action Plan
Define and document the required action steps for correcting the issue now and preventing it in the future. Once documented, assign roles and responsibilities to your team and conduct the action steps. Common actions include updating procedures, reworking a process, adding inspections, training, supplier changes, manufacturing or storage changes, policy changes, etc.
“All of these actions should be well-documented and managed by the CAPA owner.”— Larry Stevens, RAC
8. Review, Approve, and Verify Effectiveness
Once finished, the Quality Manager, or ideally, the QRB conducts a review to ensure the process was completed exactly as described. Once reviewed, the Quality Manager or QRB certifies the CAPA was carried out correctly. Then, a set of requirements must be established to prove that the CAPA plan was effective at correcting and preventing the problem. Lastly, verification requirements must be validated to ensure they were met and the issue was resolved.
“This is something FDA looks at in determining the efficiency and effectiveness of your CAPA program. Are CAPAs reviewed on a regular basis and are they closed in a reasonable amount of time? Now, occasionally, a CAPA may extend to weeks or months for the effectiveness check. So, maybe there’s a provisional closure pending the completion of that check—showing everything you needed to do is essentially done.”— Larry Stevens, RAC
Common CAPA Problems to Avoid
- Importing the 8D problem-solving model: You’ll notice that the process laid out above positions verification after implementing corrective and preventive actions. This is often found to be flipped in CAPA procedures, specifically when the process is derived from the Eight Disciplines (8D) methodology. While this system is commonly used among quality engineers in the automotive industry, its import into the device world puts it at odds with regulatory demands both under FDA and ISO regulations. In short, verification of effectiveness must be conducted after those actions are implemented.
- Unreasonable timeframes and deadlines: Another common problem is a tendency to use short, arbitrary timeframes for completing activities in an effort to convey a sense of tight control. While the logic here is simple—tight control looks compliant—timeframes set in a vacuum are a recipe for disaster when they can’t be met. In this case, a procedure that can’t be carried out as written can be even worse than not having a procedure in the first place. By design, it’s set up to fail.
- Feedback loops: Sources of information are incredibly important in just about any procedure. Misaligned inputs mean missing information. To ensure your inputs are aligned, identify the various processes that have worked for revealing root causes in the past. Discuss these matters with each of the stakeholders and write out a process in flow chart form. With this in hand, a procedure can be written or updated with verbiage, steps, and sections for responsibilities, purpose, scope, etc.
- Political influence: Problems caused by political dynamics can be particularly insidious and difficult to address within an organization. While endless white papers could be written on the effects of ego, power, and personalities, detecting their influence on your CAPA program is often relatively simple: Are too many people touching it? Overcommitting staff in the approval process, in particular, can be a clear signal that problems may be getting swept under the rug—or conversely—the program is being overused. Politics are also usually at play when viable solutions get turned down for a myriad of reasons. In general, the remedy is having the right people—and the right number of people—at each level. If one step feels bloated, it typically is.
The Value an Expert Third-Party Consultant Can Bring to Your CAPA Program
We’d like to leave you with a well-stated perspective on the value of partnering with quality systems experts when establishing or improving processes, procedures, and more.
“I’ve spent thirty years working in the industry and there’s nothing in medical device manufacturing quality systems I haven’t touched one way or another—most of it many times over. When it comes to the value of an effective, knowledgeable consultant, there’s real fresh perspective, untainted by the, ‘we don’t do it that way here.’ Consultants come in from the outside and they’ve seen a million ways to do it. They’re adaptable to help you develop a process that best fits that organization, rather than just something that’s been 'bandaid-ed' over the years and never really was made efficient. I’m overusing the word politics, but and they come in with no politics. The consultant has no ax to grind with any department. You don’t have any fighting between, or power struggles, between functions because the consultants coming in, and he just wants to give them the best answer.” — Brian Dense
4 Ways Regulatory Compliance & Quality Assurance Consultants/Contractors Offer Unique Value and Cost Effectiveness
While some circumstances clearly underscore the need for outside expertise, the value of engaging a consultant, both financially and operationally, may not always be apparent. To avoid wasted time and budget—and potentially serious quality and compliance problems that could otherwise be avoided with the help of an experienced consultant, we’ve summarized a few of the major ways companies realize the benefits of bringing in an expert.
1. Answering complex regulatory questions and navigating new regulatory territory
While new regulatory challenges impact both drug and device companies, the device industry, in particular, is contending with sweeping new requirements on top of an increasing number of combination products that present complex regulatory issues of their own (regulatory authority jurisdictions, pre-market submission types, clinical trial design, multi-center review, etc.).
Third-party experts who specialize in specific domains are very often relied upon to step in and help internal teams navigate these complex areas.
The benefit, in this case, isn’t lost when the consultants leave. In addition to leading audits and conducting remediation and revision projects to bring a company into compliance with new requirements, these individuals offer unique and often indispensable training resources that give internal teams the insight needed to implement and maintain new processes, procedures, and systems well after the consultant’s work is done.
2. Preventing the costs of enforcement and its expensive aftermath
Increased regulatory compliance enforcement over the past few years has led to an explosion of new codes while underscoring the importance of putting effective compliance safeguards in place. The costs of these preventive measures are almost always far less than the expensive consequences of enforcement action.
In addition to the disruption and costs of an investigation itself, resulting settlements, subsequent multi-year reviews, effectiveness audits, and related litigation from shareholders and plaintiffs can all lead to massive combined expenses that pale in comparison to effective prevention measures.
Third-party consultants can provide objective assessments through robust quality system auditing to evaluate the key areas that come under regulatory scrutiny in areas such as GMP, GCP, GLP, Vendor/Supplier Management, Pharmacovigilance, and Data Integrity. These measures, when conducted routinely by unbiased outside experts, can be invaluable in preventing quality system issues from developing into system-wide problems that can have enormous implications, both financially and otherwise.
3. Avoiding the practical challenges of hiring from a small talent pool
For most small or medium-sized companies, hiring high-salary internal regulatory compliance staff is simply unfeasible from a business perspective. Even for large firms, finding the top talent to fill these positions can be incredibly challenging. The relatively small pool of QA/RA talent creates intense competition for skilled personnel.
With the regulatory environment becoming increasingly globalized, it can be difficult for in-house regulatory departments to keep up with various regulatory requirements and changes. Third-party experts can achieve harmony between disparate sets of requirements by conducting integrated regulatory assessments against national requirements from multiple jurisdictions.
4. Bringing industry best practices and standards to your processes, procedures, and systems
Third-party consultants encounter a variety of challenges working in the field, and thus, develop a variety of solutions to address them. This gives them a very unique perspective into what works best when addressing a certain problem or preparing for new development.
In general, most companies like to know what other companies are doing to handle similar issues. While consultants are obviously bound by contract not to reveal specifics, they’re expected to use their experience when developing best practices and standards to bring to each organization they work while also adding the scale and bandwidth needed to augment internal teams.
Consider the Costs
For many companies, seeking outside help from third-party experts can provide overall greater value compared to doing all regulatory work internally. While per-hour billing rates may initially appear high, the total financial picture can be one of cost savings when consultants are able to provide an ROI on overall efficiency versus the alternative.
- Effective consultants are valued for their ability to complete work quickly and without rework. This factor on its own is often enough to realize cost savings. Consultants who have conducted the same work many times and aren’t affected by the distractions of daily tasks that internal personnel are can often perform tasks far faster. Of course, consultants can only perform fast, effective work when the goals of the project are well-defined and organized ahead of time. This, in addition to ensuring a consultant is hired for their expertise rather than their billing rate are the two major factors that maximize efficiency while getting achieving the goals of a project.
- Companies can lower overall costs by carefully choosing the types of projects and processes that they outsource. While just about any situation can warrant the added assistance of a consultant, consider those that require a high degree of specialization and/or those that are conducted on a periodic or project-based frequency. As FDA-regulated manufacturers continue to compete for top talent in a globalized market while navigating a more complex regulatory environment, regulatory compliance and quality assurance consultants will continue to offer valuable pathways for planning and completing projects quickly, reliably, and cost-effectively.
Join the thousands of companies executing their projects with The FDA Group.
The FDA Groups helps life science organizations rapidly access the industry's best consultants, contractors, and candidates. Our resources assist in every stage of the product lifecycle, from clinical development to commercialization, with a focus in Quality Assurance, Regulatory Affairs, and Clinical Operations.
Our auditors can perform a detailed assessment of your existing quality systems and processes to highlight problem areas, and recommend and optionally implement improvements to build quality systems that are appropriate for your company’s stage of development. We can assist you with all aspects of compliance as they impact your product, including GLP, GMP, QSR, and GCP.
The FDA Group also specializes in planning and conducting comprehensive remediation projects, Our team of former FDA and industry professionals works hand-in-hand with regulated manufacturers to uncover the root cause of compliance issues, remediate them, and implement the necessary measures to safeguard your reputation for quality both now and in the future.
With over 2,500 resources worldwide, over 225 of whom are former FDA, we meet your precise resourcing needs through a fast, convenient talent selection process supported by a Total Quality Guarantee. Get direct access to the specialized life science talent you need to bring products to market and keep them there. Whether you’re looking for consulting or project support, full-time contract, contract-to-hire, or direct-hire talent, we rapidly identify the right resource the first time.
Learn more about our services by functional area:
- Quality Assurance
- Regulatory Affairs
- Clinical Operations
- Commissioning, Qualification, and Validation
- Chemistry, Manufacturing, and Controls (CMC)
Learn more about our services by engagement model:
Get in touch with us to learn more and get the support you need—when and where you need it.
FREE WHITE PAPER
The Guide to CAPA & Root Cause Analysis in FDA-Regulated Industries
Download the full white paper and take this guide with you. It offers everything we cover here, and more, in a handy PDF you can use to assess and enhance your CAPA program.