
Meeting regulatory and quality compliance in multiple markets is a difficult and complex challenge.
Manufacturers must comply with strict regulatory requirements and undergo regular quality system audits, which can interfere with daily operations. Auditors often duplicate efforts when evaluating approved systems.
The Medical Device Single Audit Program (MDSAP) simplifies compliance with global quality management system (QMS) standards and reduces the industry's resource burden.
MDSAP Basics: A Brief Review
What is MDSAP?
MDSAP is a somewhat new program headed by the International Medical Device Regulators Forum. It was launched as a pilot program in 2012 through 2013 and was fully implemented in 2016 after a successful trial.
The program was established by the Global Harmonization Task Force, which comprises of international medical regulators who aimed to streamline the requirements for getting QMS certifications for international companies.
This single audit program has two primary objectives:
- Avoid duplication of regulatory resources across multiple countries.
- Minimize disruptions of medical device manufacturers from multiple regulatory audits.
MDSAP aims to create a global coalition for medical device manufacturing oversight, reducing regulatory burden and business interruptions. A reliable, universally accepted audit program, the idea goes, would save costs and reduce multiple audits.
How does MDSAP eligibility work?
Manufacturers from anywhere in the world have the opportunity to take part in the MDSAP. Any manufacturer can participate as long as a product falls under the jurisdiction of at least one participating Regulatory Authority and meets its quality management system requirements.
It's important to note that only the countries participating in MDSAP will have direct access to the audit reports.
How does MDSAP work in practice?
The general concept of the MDSAP can be thought of as a dynamic relationship between manufacturers (Mfr), regulatory authorities (RA), and auditing organizations (AO) as shown in the diagram below.
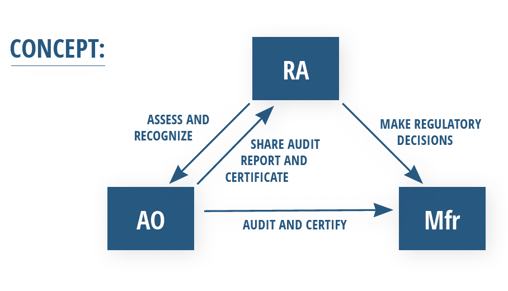
Official MDSAP audits are conducted by private AOs that are qualified to meet MDSAP auditor requirements. Manufacturers can hire an AO to perform MDSAP audits, and often, these AOs can also perform ISO 13485 audits simultaneously.
The MDSAP audit process encompasses seven key process elements:
- Management
- Device marketing authorization and facility registration
- Medical device adverse events and advisory notices reporting
- Measurement, analysis, and improvement
- Design and development
- Production and service controls
- Purchasing
During a MDSAP audit, the impact of risk management on various processes is carefully analyzed. To ensure preparedness, each section has specific questions that must be answered. This allows manufacturers to anticipate the auditor's expectations. The actual audit takes a process-by-process approach, with some interconnections, which ensures that the auditor and client understand the entire MDSAP audit process.
- Each task is assigned a specific amount of time, typically ranging from 15 to 44 minutes, based on its complexity.
- The audit duration is adjusted depending on whether sterilization, service, installation, implants, or design are involved. For example, these tasks can take up to 45 minutes for each, while design may require five hours.
- Additionally, the duration is extended for critical supplier visits, which can take up to four hours, and for addressing outstanding nonconformance reports.
The MDSAP audit renewal timeline closely aligns with ISO 13485. Once certified, the validity of the certification lasts for three years, starting with an initial audit. Throughout that period, two surveillance audits and a recertification audit must be conducted. This ensures that your organization maintains compliance and meets the expectations set by the MDSAP program.
The MDSAP audit sequence
Diving into the details of how these auditing and assessment activities work, the MDSAP operates on a three-year cycle involving four separate audits:
- An initial assessment
- Two surveillance assessments
- A final re-recognition assessment
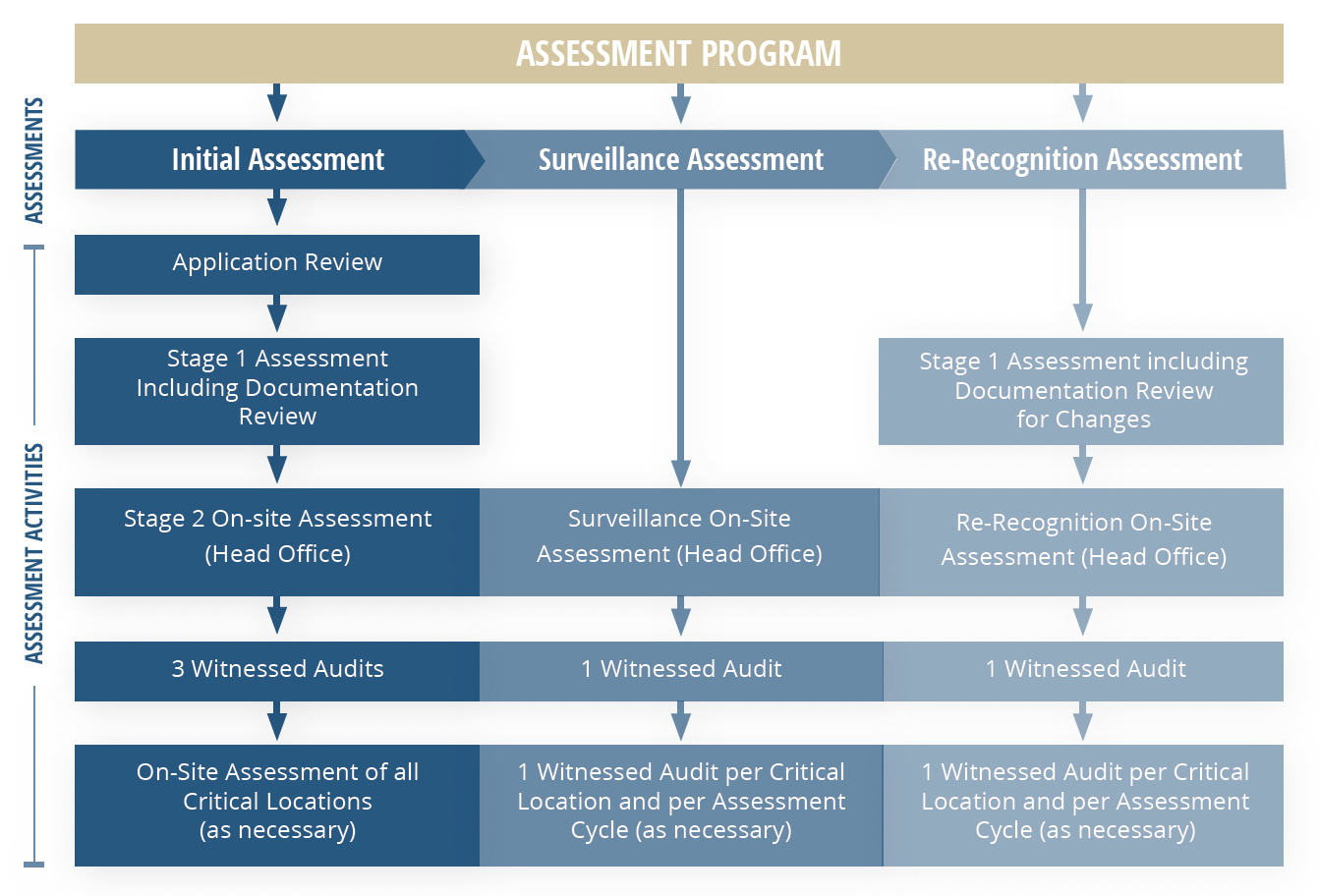
Let's break this audit sequence down:
The initial assessment
- Stage 1: Following application review, much like a standard ISO certification, the MDSAP assessment program begins with a review of impacted documentation to determine preparedness for an on-site assessment of both the head office and any critical locations.
- Stage 2: Once the Notified Body is satisfied with your documentation and overall preparedness, they will conduct an on-site audit to evaluate quality management system (QMS) compliance to ISO 13485 and any other requirements of MDSAP-participating regulatory authorities. Expect a full quality system review during this process. The MDSAP Companion Document is a useful resource explaining the audit questions to expect and other important considerations for each region. This document also highlights the subtle differences between current FDA and ISO concepts and requirements to help you prepare accordingly. Treat this like your MDSAP handbook.
Surveillance assessments
Two Surveillance Audits are conducted in years one and two to assess any changes made to your products or QMS processes since the initial assessment was completed. Typically, these audits are structured so that each half of your system is assessed with each audit.
Following the second audit, your Notified Body will review any findings observed during the prior audits. Theoretically, these should be cheaper and shorter; however, in this early phase of the program, this still remains to be seen.
Re-recognition assessment
While technically included in the MDSAP audit cycle, a Re-Recognition Assessment is actually conducted in year four. This audit will evaluate your QMS for continued suitability and effectiveness in meeting QMS requirements under the MDSAP.
Typically, this and the prior Surveillance Assessments should not include an in-depth evaluation of procedures unless significant changes have been made to your quality system, facility, or products, such as moving from one device class to another or after adding manufacturing operations to what was solely a design-based organization.
In situations like these, your Notified Body will likely evaluate any impacted documentation and associated procedures.
MDSAP's point-based nonconformity grading system
One of the most significant new concepts the MDSAP brings is its point-based nonconformity grading system. To make nonconformity grading more consistent, the MDSAP does away with traditional grading criteria such as “significant finding,” “regular finding,” and “significant opportunity for improvement,” and instead uses a point system described at length in the Global Harmonization Task Force (GHTF)’s Quality management system - Medical devices - Nonconformity Grading System for Regulatory Purposes and Information Exchange.
We’ve summarized this complex reference guide in simpler terms below. The grading system works in two steps. First by assigning points to nonconformities written against requirements in ISO 13485:2016 using a 4-point grading matrix and then applying that initial point score to a series of escalation rules that may or may not result in a higher final point grade.
Let’s explore the key concepts for both steps.
Step one: the grading matrix
Point grades in step one are determined using the grading matrix below. This divides the clauses of ISO 13485:2016 into two categories: those that indirectly impact the QMS and those that directly impact the QMS.
- “Indirect QMS impact” — encompassing clauses 4.1 through 6 — is categorized as the “administrative enabler” section of ISO 13485:2016. These requirements make it possible or feasible for QMS processes to operate and are considered to indirectly influence medical device safety and performance.
- “Direct QMS impact” — encompassing clauses 6.4 through 8.5 — is the category of requirements that have a direct influence on design and manufacturing controls, which in turn, directly impacts product safety and performance.
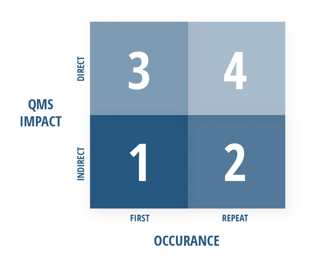
The occurrence of the nonconformity also plays a factor in the grading system, as illustrated in the visual matrix above. The details of what constitutes a first or repeat occurrence can get somewhat complicated, so we’ve summarized the key concepts in the simplest terms possible:
- A “first” occurrence is a nonconformity in a particular sub-clause (X.X.X) of ISO 13485:2016 that has not been observed in the two previous QMS audits which evaluated the same sub-clause.
- A “repeat” occurrence is a nonconformity that has been identified within either of the two previous QMS audits that evaluated that same sub-clause (X.X.X). These have been determined to pose a higher risk because they indicate corrective action hasn’t been adequately taken or implemented, and as such, receives a higher (or worse) grade.
It’s important to note that an “occurrence” is directed at the frequency of a nonconformity cited from one audit to the next performed by the same auditing organization. It is not the occurrences of examples within a given sample size that the auditor may take to determine if a nonconformity exists during an audit.
Since nonconformities can be written up against more than one clause, it’s up to the auditor to determine the impact of the nonconformity on the QMS and assign the appropriate clause. The GHTF document offers helpful examples that illustrate how grading works in this initial step.
Step two: grading escalation rules
Once a point grade (1 through 4) is determined using the matrix, it’s then subjected to an escalation process to address areas of higher risk that could affect product safety and performance. The grade determined by step one is increased by 1 point for each rule that applies in the...
Absence of a documented process or procedure. (In this case, a process or procedure is “absent” when it hasn’t been documented for the requirement.)
or
Release of a Nonconforming Medical Device. (If a nonconformity resulted in releasing a nonconforming medical device to the market, it’s direct evidence of a QMS failure. However, if a nonconforming device is released with adequate technical and scientific justification, the nonconformity is considered to be resolved, and the rule will not apply.)
The final nonconformity grade will be between 1 and 6, however, a grade of 5 or above is determined to carry a high enough risk that intervention is required. Grades of 6 will be listed as 5 since the differentiation between the two scores offers no benefit within the grading system.
Scores will be recorded on the standardized Regulatory Audit Information Exchange Form, which offers a common means of exchanging audit information between regulatory authorities. This form will be given to manufacturers following MDSAP audits during the closing meeting.
It’s important to note that grades assigned to nonconformities should not be changed following any corrective actions taken by the manufacturer, however, they may be amended on the conditions of the auditing organization’s appeals process.
Which countries participate in MDSAP?
At present, MDSAP has the active participation of five countries, each of which includes specific market requirements within the framework of this single audit program. Although the European Union currently holds the status of an "Official Observer," it is not a member of MDSAP.
- United States (FDA 21 CFR 820) - FDA will accept MDSAP audit results instead of routine inspections for medical device companies. However, initial visits and "for cause" inspections will still be conducted.
- Health Canada (CMDR) - As of January 1st, 2019, Health Canada only accepts MDSAP audits from medical device manufacturers following a successful three-year pilot from 2014-2016.
- Brazil (ANVISA RDC 16) - Brazilian authorities will accept MDSAP for initial audits, but will still conduct their own inspections for higher-risk devices.
- Japan (MHLW Ministerial Ordinance No. 169) - Japanese authorities now accept MDSAP audit results instead of on-site J-QMS audits.
- Australia (TGA) - The Australian Therapeutic Goods Administration (TGA) now recognizes that passing an MDSAP audit satisfies QMS requirements. MDSAP certificates are equivalent to CE certificates.
What are the benefits of MDSAP?
There are several genuine advantages of participating in MDSAP:
- Time efficiency: Manufacturers save significant time as MDSAP consolidates multiple audits into one. Preparation, responses, and remediation are streamlined, freeing up time for other essential tasks.
- Cost reduction: Single audits are cost-effective, eliminating the financial strain of multiple separate audits.
- Predictable auditing schedule: MDSAP provides a calculable audit schedule, aiding in efficient resource allocation. The audit calculator offers precise time estimates based on the specific medical device.
- Clear and prescriptive process: The program outlines a clear, step-by-step process, reducing uncertainties and queries. Manufacturers can tailor their Quality Management Systems (QMS) to MDSAP’s specific requirements, enhancing success rates.
- International certification: MDSAP certification is mandatory in Canada and is recognized by four other countries, reducing the need for additional audits. Manufacturers often certify to all participating countries, optimizing time and resource utilization.
- Graded Nonconformity Findings: Nonconformities are graded by severity, enabling prioritized and efficient remediation efforts.
- Scaled Audits for Larger Companies: Audits are scaled based on the QMS, not the number of employees or products, often leading to shorter audit durations. Also, the audit duration is determined by the scope, not the company size, aligning with ISO 13485 standards.
Regulators and industry also likes MDSAP because lowers the regulatory burden and accelerates the process of getting medical devices to patients across multiple markets.
An Overview of the MSDSAP Audit Approach
The MDSAP audit approach is explained in full detail in MDSAP's 224-page guide.
The audit sequence is designed to be flexible, allowing for adjustments to ensure an efficient and effective audit. Auditors can make justified exceptions to the sequence to optimize audit time and planning, ensuring core elements and risk-based sample selections are respected.
Here's a top-line overview of each broad component of an audit.
1. Management
(Page 19) The management process audit ensures that internal audits are conducted according to established procedures by trained individuals. It checks the compliance and effectiveness of quality management system (QMS) procedures. The audit team confirms that the output of internal audits is an input to management review, ensuring a loop of continuous improvement.
2. Measurement, Analysis, and Improvement
(Page 51) Auditors review the Measurement, Analysis, and Improvement process early in the audit. They focus on corrective actions resulting from design changes or product nonconformities attributed to design. The audit team considers selecting designs for review, especially those related to identified quality problems.
3. Device Marketing Authorization and Facility Registration
(Page 37) This part of the audit ensures that the organization has followed proper channels to obtain authorization for device marketing and facility registration. It also checks for evidence of marketing clearance or approval and the notification process for changes to marketed devices or the QMS.
4. Medical Device Adverse Events and Advisory Notices Reporting
(Page 77) The audit examines the organization’s process for notifying adverse events and advisory notices. It ensures that the procedures are in place and are followed to report incidents effectively and comply with regulatory requirements.
5. Design and Development
(Page 89) Auditors review the design and development planning, focusing on addressing corrective actions and product nonconformities. They pay special attention to the relationship between identified quality problems and specific aspects of design and development, ensuring that risk control methods are effective.
6. Production and Service Controls
(Page 116) The audit of this section begins after gathering enough information from the review of core elements in previous sections. It ensures that the production and service processes are well-controlled, monitored, and measured for product conformity.
7. Purchasing
(Page 157) Auditors assess the planning activities regarding purchased products and outsourced processes. They review the selection and evaluation of suppliers, verification of purchased products, and the top management's commitment to the purchasing process.
8. Special Audits
(Page 17) These are extraordinary audits not part of the planned audit cycle, used when necessary. They focus on specific elements of the organization’s QMS and may include audits conducted in response to an application for an extension to the scope of certification.
Annexes
The annexes provide additional detailed information and guidance on specific audit areas, including product/process-related technologies, requirements for sterile medical devices, and adverse events and advisory notices reporting process.
A Few MDSAP Frequently Asked Questions
What is the MDSAP?
In 2012, the International Medical Device Regulators Forum (IMDRF) initiated the Medical Devices Single Audit Program (MDSAP) to streamline global auditing of medical device manufacturers for enhanced safety. Manufacturers can hire an Auditing Organization to evaluate their Quality Management System annually, aligning with regulations in Australia, Brazil, Canada, Japan, and the U.S. The MDSAP reduces the frequency of audits, with each country interpreting the results based on its laws. Currently, only Canada mandates MDSAP participation for certain classes of medical devices. Audit results are stored in a shared database accessible to all member countries' regulatory authorities.
Is MDSAP participation mandatory?
Participation in the MDSAP is optional in Australia, Brazil, Japan, and the United States, but it's mandatory in Canada for class II, III, and IV medical devices since January 1, 2019, as per the details on the Health Canada website. Manufacturers opting for MDSAP must specify the countries where they market or plan to market their devices in the certification contract. The on-site audit evaluates the manufacturer’s QMS compliance against the regulations of the listed countries.
What is the MDSAP beneficial?
The MDSAP is a global initiative that unifies quality management system audits for enhanced medical device safety worldwide. It benefits manufacturers aiming to market in its five core countries and two new affiliates, South Korea and Argentina. Participants gain streamlined regulatory approval in these nations and ISO 13485:2016 certification for QMS compliance.
Is MDSAP participation mandatory?
Participation in the MDSAP is optional in Australia, Brazil, Japan, and the United States, but it's mandatory in Canada for class II, III, and IV medical devices since January 1, 2019, as per the details on the Health Canada website. Manufacturers opting for MDSAP must specify the countries where they market or plan to market their devices in the certification contract. The on-site audit evaluates the manufacturer’s QMS compliance against the regulations of the listed countries.
How are the MDSAP audit criteria aligned with ISO 13485 provisions?
The MDSAP audit criteria encompass the provisions of ISO 13485 and additional requirements from participating regulatory authorities. Manufacturers can tailor the criteria to exclude regulations of countries where they don't intend to market their devices. Thus, the audit criteria combine ISO 13485 and specific regulatory requirements of the countries where the manufacturer operates or plans to enter.
How The FDA Group Can Help
If your company is participating or considering participating in the MDSAP, our experienced auditing professionals can assess your current processes against the MDSAP requirements and help you identify and remediate key areas while training staff to maintain compliance under MDSAP. As an auditing and remediation resourcing organization independent of certification or regulatory bodies, this preparatory audit enables you to assess and remediate your system confidentially.
Our experienced MDSAP auditors use the same MDSAP process-based audit model that the Auditing Organizations (AO) will use to calculate your MDSAP audit score and identify your areas of biggest risk. Many clients choose to include an assessment of their quality system against ISO 13485:2016 and the MDSAP requirements to allow for a more comprehensive evaluation.
We recognize the challenges of developing and managing an effective quality system under the watchful eye of regulators. Our quality system resourcing professionals draw on years of experience and highly specialized skills to ensure objective, accurate, and actionable recommendations are made each step of the way.
Our proprietary talent selection process brings together a diverse array of skill sets, experience, and expertise to offer a truly unique opportunity for companies interested in receiving personal and professional attention.
Contact us today to learn more about our MDSAP auditing and support services.

FREE CASE STUDY
Deviation Backlog Reduction, CAPA Management, and Long-Term Staff Augmentation
See how we successfully partnered with a multinational pharmaceutical company to address deviation and CAPA backlogs, implement corrective actions, provide staff training, and achieve the goal of eliminating all backlogs within the allocated time and budget.
